MACE-POLAR-1: A Polarisable Electrostatic Foundation Model for Molecular Chemistry
作者: Ilyes Batatia, William J. Baldwin, Domantas Kuryla, Joseph Hart, Elliott Kasoar, Alin M. Elena, Harry Moore, Mikołaj J. Gawkowski, Benjamin X. Shi, Venkat Kapil, Panagiotis Kourtis, Ioan-Bogdan Magdău, Gábor Csányi
分类: physics.chem-ph, cs.LG
发布日期: 2026-02-23
💡 一句话要点
MACE-POLAR-1:用于分子化学的可极化静电基础模型
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 分子化学 静电相互作用 机器学习原子间势 长程静电效应 可极化模型
📋 核心要点
- 现有MLIPs依赖局部原子描述符,无法捕捉长程静电效应,限制了对分子间相互作用的准确建模。
- MACE-POLAR-1结合局部几何特征与非自洽场方法,通过可极化迭代更新电荷和自旋密度,模拟静电感应。
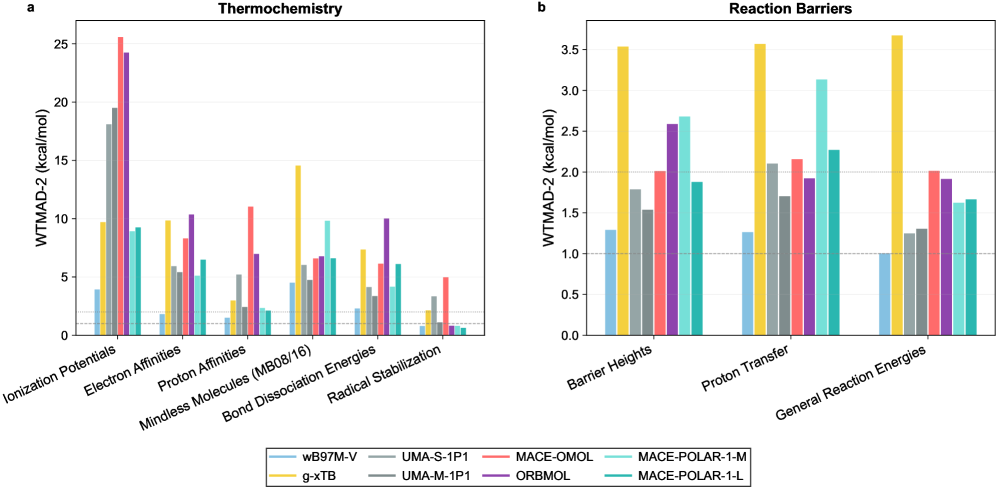
- 实验表明,该模型在热化学、反应势垒等方面达到化学精度,显著提升了对非共价相互作用的描述。
📝 摘要(中文)
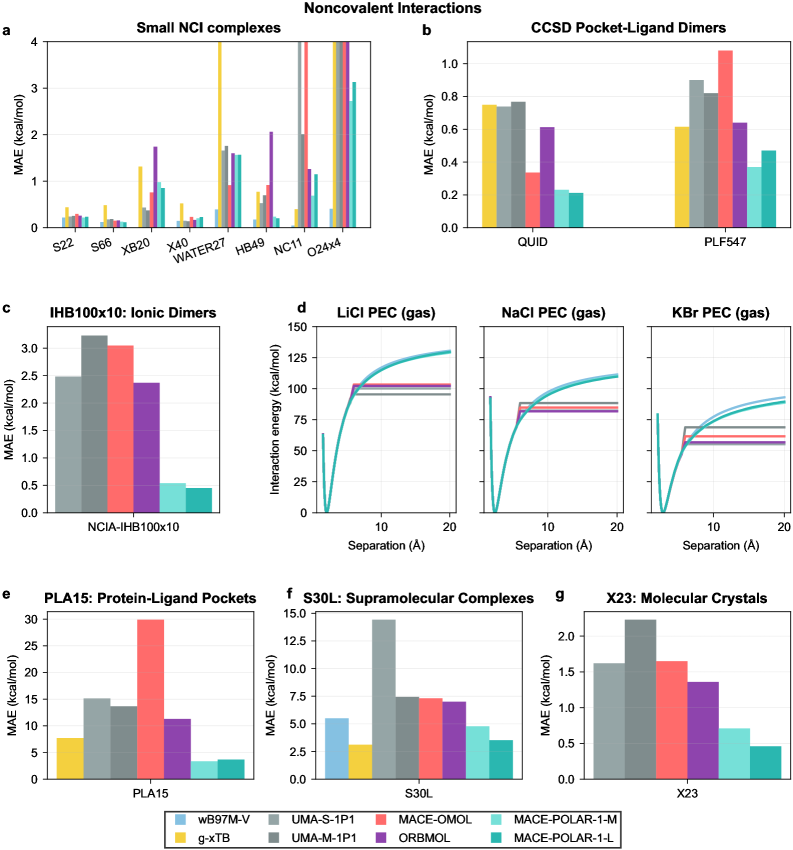
精确建模静电相互作用和电荷转移是计算化学的基础,但大多数机器学习原子间势(MLIPs)依赖于无法捕捉长程静电效应的局部原子描述符。我们提出了一种新的分子化学静电基础模型,该模型扩展了MACE架构,显式处理长程相互作用和静电感应。我们的方法将局部多体几何特征与非自洽场形式相结合,通过可极化迭代更新可学习的电荷和自旋密度来模拟感应,然后通过可学习的福井函数进行全局电荷平衡,以控制总电荷和总自旋。这种设计能够准确且物理地描述具有不同电荷和自旋状态的系统,同时保持计算效率。我们的模型在包含1亿次混合DFT计算的OMol25数据集上进行训练,在各种基准测试中实现了化学精度,在热化学、反应势垒、构象能量和过渡金属配合物方面的精度与混合DFT相当。值得注意的是,我们证明了包含长程静电效应可以显著改善对非共价相互作用和超分子配合物的描述,在X23-DMC数据集中实现了亚千卡/摩尔的分子晶体形成能预测,并且在蛋白质-配体相互作用方面比短程模型提高了四倍。该模型能够处理可变的电荷和自旋状态,响应外部场,提供可解释的自旋分辨电荷密度,并保持从小分子到蛋白质-配体配合物的精度,使其成为计算分子化学和药物发现的多功能工具。
🔬 方法详解
问题定义:现有机器学习原子间势(MLIPs)在模拟分子体系时,难以准确捕捉长程静电相互作用和电荷转移。这些模型通常依赖于局部原子描述符,忽略了分子间静电效应,导致在非共价相互作用、超分子体系以及具有不同电荷和自旋状态的体系中表现不佳。现有方法的痛点在于缺乏对长程静电效应的显式建模,限制了其在复杂分子体系中的应用。
核心思路:MACE-POLAR-1的核心思路是将局部多体几何特征与非自洽场(non-self-consistent field)方法相结合,显式地处理长程静电相互作用和静电感应。通过可极化迭代更新可学习的电荷和自旋密度,模拟静电感应过程,并利用可学习的福井函数进行全局电荷平衡,从而控制总电荷和总自旋。这种设计旨在提供一种准确且物理的描述,能够处理具有不同电荷和自旋状态的系统,同时保持计算效率。
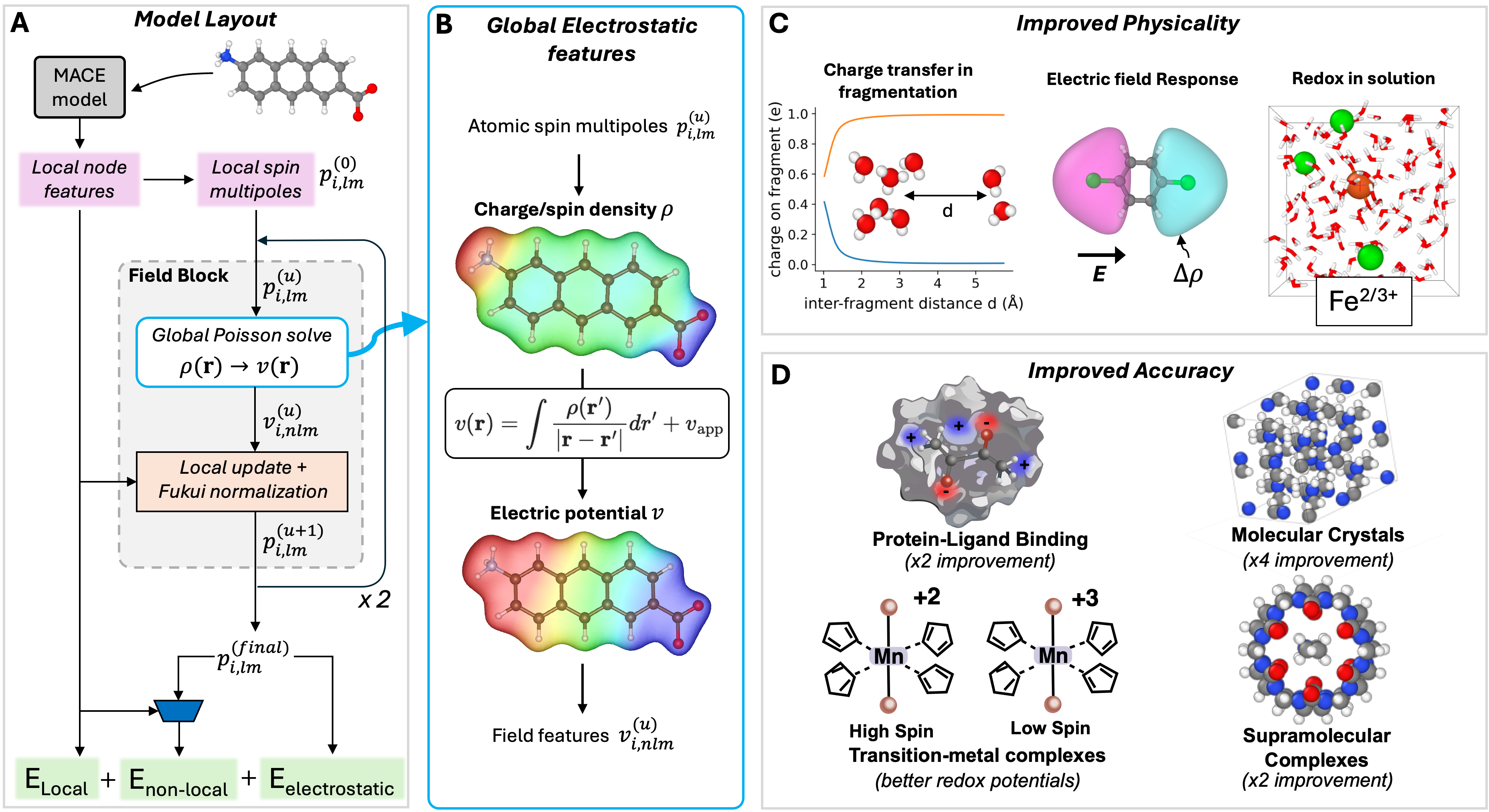
技术框架:MACE-POLAR-1的整体架构包括以下几个主要模块:1) 局部多体几何特征提取模块,用于描述原子周围的局部环境;2) 可极化迭代模块,通过非自洽场方法更新可学习的电荷和自旋密度,模拟静电感应;3) 全局电荷平衡模块,利用可学习的福井函数控制总电荷和总自旋;4) 能量预测模块,基于上述模块的输出预测体系的能量。整个流程通过迭代优化,使得模型能够更好地捕捉长程静电相互作用。
关键创新:MACE-POLAR-1最重要的技术创新点在于显式地建模了长程静电相互作用和静电感应。与传统的基于局部原子描述符的MLIPs相比,该模型能够更准确地描述分子间的静电效应,特别是在非共价相互作用和超分子体系中。此外,通过可学习的电荷和自旋密度,模型能够提供可解释的物理信息,例如原子电荷分布和自旋极化。
关键设计:在可极化迭代模块中,采用了非自洽场方法,通过迭代更新电荷和自旋密度来模拟静电感应。福井函数被设计为可学习的参数,用于控制全局电荷平衡,从而保证模型在处理具有不同电荷状态的体系时具有良好的泛化能力。损失函数的设计考虑了能量、力和电荷的准确性,通过加权的方式平衡不同性质的预测误差。
🖼️ 关键图片
📊 实验亮点
MACE-POLAR-1在多个基准测试中表现出色,在热化学、反应势垒、构象能量和过渡金属配合物方面的精度与混合DFT相当。在X23-DMC数据集中,该模型实现了亚千卡/摩尔的分子晶体形成能预测。在蛋白质-配体相互作用方面,该模型比短程模型提高了四倍。这些结果表明,MACE-POLAR-1能够显著提升对非共价相互作用和超分子配合物的描述。
🎯 应用场景
MACE-POLAR-1在计算分子化学和药物发现领域具有广泛的应用前景。它可以用于准确预测分子性质、模拟化学反应、优化分子结构以及筛选药物候选分子。该模型能够处理具有不同电荷和自旋状态的体系,并提供可解释的物理信息,使其成为研究复杂分子体系和设计新型功能材料的有力工具。未来,该模型有望加速药物研发进程,并推动计算化学的发展。
📄 摘要(原文)
Accurate modelling of electrostatic interactions and charge transfer is fundamental to computational chemistry, yet most machine learning interatomic potentials (MLIPs) rely on local atomic descriptors that cannot capture long-range electrostatic effects. We present a new electrostatic foundation model for molecular chemistry that extends the MACE architecture with explicit treatment of long-range interactions and electrostatic induction. Our approach combines local many-body geometric features with a non-self-consistent field formalism that updates learnable charge and spin densities through polarisable iterations to model induction, followed by global charge equilibration via learnable Fukui functions to control total charge and total spin. This design enables an accurate and physical description of systems with varying charge and spin states while maintaining computational efficiency. Trained on the OMol25 dataset of 100 million hybrid DFT calculations, our models achieve chemical accuracy across diverse benchmarks, with accuracy competitive with hybrid DFT on thermochemistry, reaction barriers, conformational energies, and transition metal complexes. Notably, we demonstrate that the inclusion of long-range electrostatics leads to a large improvement in the description of non-covalent interactions and supramolecular complexes over non-electrostatic models, including sub-kcal/mol prediction of molecular crystal formation energy in the X23-DMC dataset and a fourfold improvement over short-ranged models on protein-ligand interactions. The model's ability to handle variable charge and spin states, respond to external fields, provide interpretable spin-resolved charge densities, and maintain accuracy from small molecules to protein-ligand complexes positions it as a versatile tool for computational molecular chemistry and drug discovery.