AdsorbFlow: energy-conditioned flow matching enables fast and realistic adsorbate placement
作者: Jiangjie Qiu, Wentao Li, Honghao Chen, Leyi Zhao, Xiaonan Wang
分类: cs.LG
发布日期: 2026-02-22
💡 一句话要点
AdsorbFlow:能量条件流匹配实现快速逼真的吸附质放置
🎯 匹配领域: 支柱二:RL算法与架构 (RL & Architecture) 支柱四:生成式动作 (Generative Motion)
关键词: 吸附质放置 流匹配 能量条件 催化剂设计 生成模型
📋 核心要点
- 计算催化中,寻找吸附质在催化剂表面的低能量吸附位点是关键瓶颈,现有方法计算成本高昂。
- AdsorbFlow通过能量条件流匹配学习吸附质的刚体配置空间,使用确定性向量场进行快速采样。
- 实验表明,AdsorbFlow在吸附质放置任务上,比现有扩散模型更快、更准确,且异常率更低。
📝 摘要(中文)
在催化表面上识别低能量吸附几何结构是计算非均相催化的一个实际瓶颈:困难不仅在于密度泛函理论(DFT)的成本,还在于提出能够松弛到正确能量盆地的初始放置。条件去噪扩散提高了成功率,但每个样本需要约100个迭代步骤。本文介绍AdsorbFlow,这是一种确定性生成模型,它通过条件流匹配学习吸附质平移和旋转的刚体配置空间上的能量条件向量场。能量信息通过无分类器指导条件输入——而不是能量梯度指导——并且采样简化为在少至5步中积分一个ODE。在具有完整DFT单点验证的OC20-Dense上,具有EquiformerV2主干的AdsorbFlow实现了61.4%的SR@10和34.1%的SR@1——在每个评估级别上都超过了AdsorbDiff(31.8%的SR@1,41.0%的SR@10)和AdsorbML(47.7%的SR@10)——同时使用少20倍的生成步骤,并实现了生成方法中最低的异常率(6.8%)。在50个分布外系统上,AdsorbFlow保持了58.0%的SR@10,MLFF到DFT的差距仅为4个百分点。这些结果表明,对于吸附质放置,确定性传输比随机去噪更快且更准确。
🔬 方法详解
问题定义:论文旨在解决计算催化中吸附质在催化剂表面吸附位点预测的问题。现有方法,如基于扩散的模型(例如AdsorbDiff),虽然取得了一定的成功,但需要大量的迭代步骤才能生成有效的吸附构型,计算成本高昂,效率较低。此外,现有方法在生成过程中可能产生不合理的结构,即异常率较高。
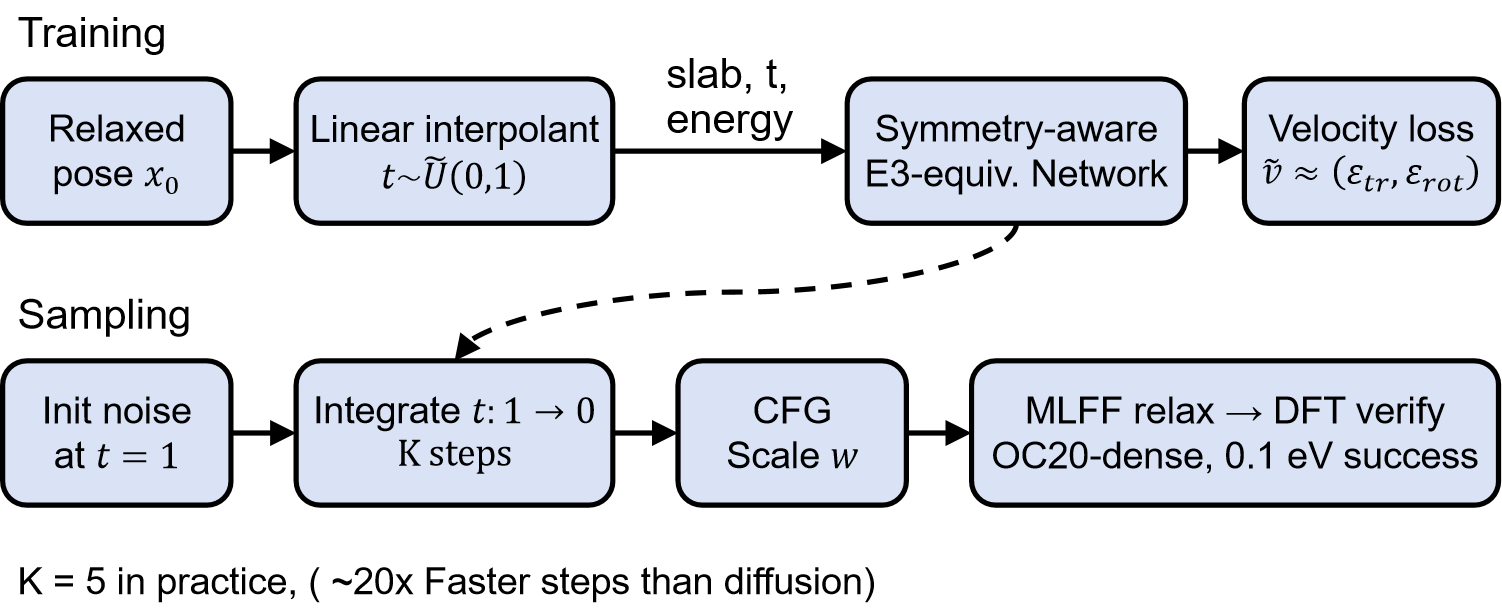
核心思路:AdsorbFlow的核心思路是利用能量条件流匹配,学习一个确定性的向量场,该向量场能够将随机初始的吸附质构型快速、准确地引导到低能量的吸附位点。通过将能量信息作为条件,模型能够学习到吸附质构型与能量之间的关系,从而实现更有效的采样。与依赖随机去噪过程的扩散模型不同,AdsorbFlow采用确定性的ODE积分,显著减少了采样所需的步骤。
技术框架:AdsorbFlow的整体框架包括以下几个主要部分:1)能量条件模块:利用无分类器指导条件(classifier-free guidance conditioning)将能量信息融入到模型中。2)流匹配模块:学习吸附质刚体配置空间上的能量条件向量场。3)ODE积分器:使用数值方法(如欧拉方法或龙格-库塔方法)对学习到的向量场进行积分,从而生成吸附质的最终构型。EquiformerV2被用作主干网络,用于提取吸附质和催化剂表面的特征。
关键创新:AdsorbFlow的关键创新在于使用确定性流匹配来替代传统的随机去噪扩散过程。这种确定性方法不仅减少了采样所需的步骤,还提高了生成构型的准确性。此外,使用无分类器指导条件而非能量梯度指导,避免了对能量梯度计算的依赖,降低了计算复杂度。
关键设计:AdsorbFlow的关键设计包括:1)能量条件方式:采用无分类器指导条件,通过联合训练一个条件模型和一个非条件模型,并在推理时通过调整它们的输出来实现能量条件的引导。2)损失函数:使用流匹配损失函数来训练模型,该损失函数旨在最小化预测向量场与真实向量场之间的差异。3)网络结构:使用EquiformerV2作为主干网络,该网络能够有效地处理原子间的相互作用,并提取具有旋转不变性的特征。
🖼️ 关键图片
📊 实验亮点
AdsorbFlow在OC20-Dense数据集上取得了显著的性能提升,SR@10达到61.4%,SR@1达到34.1%,超越了AdsorbDiff和AdsorbML等基线方法。更重要的是,AdsorbFlow仅需5步即可完成采样,比AdsorbDiff快20倍。同时,AdsorbFlow的异常率仅为6.8%,是所有生成方法中最低的。在分布外数据集上,AdsorbFlow仍保持了较高的性能,SR@10达到58.0%,MLFF到DFT的差距仅为4个百分点。
🎯 应用场景
AdsorbFlow在催化剂设计、材料科学等领域具有广泛的应用前景。它可以加速新型催化剂的发现和优化,降低实验成本。通过快速、准确地预测吸附质在催化剂表面的吸附位点,研究人员可以更好地理解催化反应机理,从而设计出更高效、更稳定的催化剂。此外,该方法还可以应用于其他涉及分子吸附的领域,如气体分离、传感器设计等。
📄 摘要(原文)
Identifying low-energy adsorption geometries on catalytic surfaces is a practical bottleneck for computational heterogeneous catalysis: the difficulty lies not only in the cost of density functional theory (DFT) but in proposing initial placements that relax into the correct energy basins. Conditional denoising diffusion has improved success rates, yet requires $\sim$100 iterative steps per sample. Here we introduce AdsorbFlow, a deterministic generative model that learns an energy-conditioned vector field on the rigid-body configuration space of adsorbate translation and rotation via conditional flow matching. Energy information enters through classifier-free guidance conditioning -- not energy-gradient guidance -- and sampling reduces to integrating an ODE in as few as 5 steps. On OC20-Dense with full DFT single-point verification, AdsorbFlow with an EquiformerV2 backbone achieves 61.4% SR@10 and 34.1% SR@1 -- surpassing AdsorbDiff (31.8% SR@1, 41.0% SR@10) at every evaluation level and AdsorbML (47.7% SR@10) -- while using 20 times fewer generative steps and achieving the lowest anomaly rate among generative methods (6.8%). On 50 out-of-distribution systems, AdsorbFlow retains 58.0% SR@10 with a MLFF-to-DFT gap of only 4~percentage points. These results establish that deterministic transport is both faster and more accurate than stochastic denoising for adsorbate placement.