Parameter-free representations outperform single-cell foundation models on downstream benchmarks
作者: Huan Souza, Pankaj Mehta
分类: q-bio.GN, cs.LG, q-bio.QM
发布日期: 2026-02-18
💡 一句话要点
提出无参数表示方法以超越单细胞基础模型的下游任务表现
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 单细胞RNA测序 线性表示 深度学习 基准测试 基因表达 生物信息学 模型评估
📋 核心要点
- 现有的单细胞基础模型如TranscriptFormer依赖于复杂的深度学习方法,计算资源消耗大且难以解释。
- 论文提出了一种基于简单线性方法的无参数表示方法,通过细致的归一化处理来捕捉细胞基因表达的特征。
- 实验结果显示,该方法在多个基准测试中表现优异,尤其在分布外任务中超越了现有的基础模型。
📝 摘要(中文)
单细胞RNA测序(scRNA-seq)数据展现出强烈且可重复的统计结构,这促使了基于变换器架构的大规模基础模型的开发,如TranscriptFormer。这些嵌入已在细胞类型分类、疾病状态预测和跨物种学习等下游任务中取得了最先进的表现。本文探讨了是否可以在不使用计算密集型深度学习表示的情况下实现类似的性能。通过依赖于细致的归一化和线性方法的简单可解释管道,我们在多个常用基准测试中获得了最先进或接近最先进的表现,尤其在涉及训练数据中缺失的新细胞类型和生物体的分布外任务中超越了基础模型。我们的发现强调了严格基准测试的必要性,并表明细胞身份的生物学可以通过单细胞基因表达数据的简单线性表示来捕捉。
🔬 方法详解
问题定义:本文旨在解决单细胞RNA测序数据分析中现有深度学习模型的计算复杂性和可解释性不足的问题。现有方法如TranscriptFormer虽然表现优异,但其计算资源消耗大,且难以理解其内部机制。
核心思路:论文提出了一种无参数的线性表示方法,旨在通过简单的归一化和线性方法来捕捉细胞基因表达的统计特征,从而实现与复杂模型相当的性能。
技术框架:整体架构包括数据预处理、归一化、线性建模和性能评估几个主要模块。首先对单细胞RNA测序数据进行归一化处理,然后应用线性模型进行特征提取,最后在多个基准测试上进行评估。
关键创新:最重要的技术创新在于提出了一种无需深度学习的线性表示方法,能够在多个下游任务中实现与深度学习模型相媲美的性能,尤其在分布外任务中表现突出。
关键设计:在方法设计中,采用了细致的归一化策略以确保数据的一致性,并通过线性回归等简单模型进行分析,避免了复杂的网络结构和参数调优过程。具体的损失函数和模型结构未在摘要中详细说明,需参考原文以获取更多细节。
🖼️ 关键图片
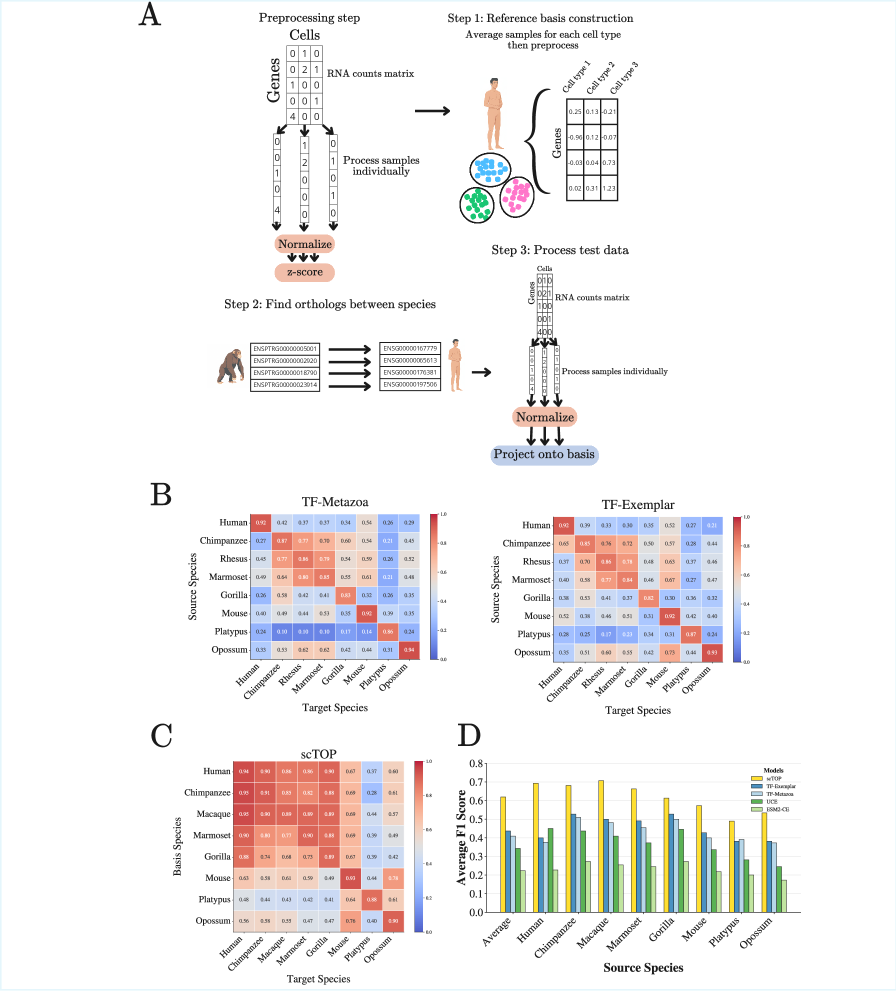
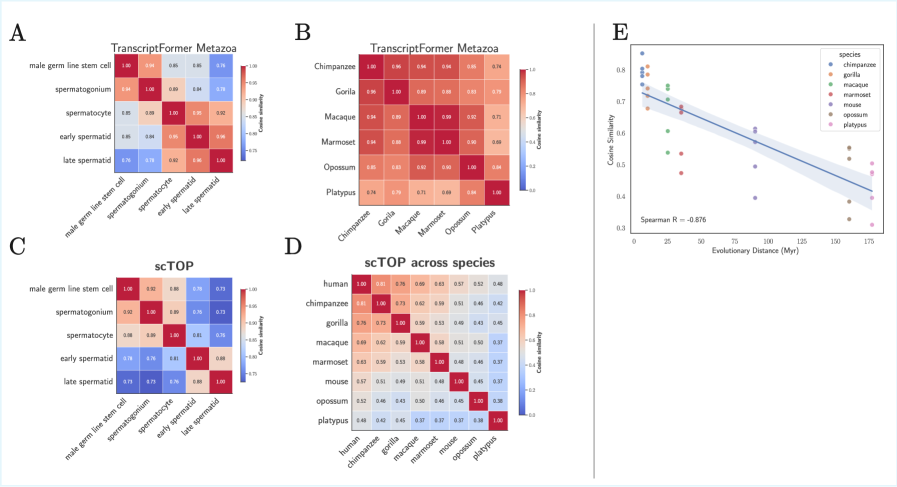
📊 实验亮点
实验结果表明,提出的无参数表示方法在多个基准测试中达到了最先进或接近最先进的表现,特别是在分布外任务中超越了现有的基础模型,显示出在新细胞类型和生物体上的优越性。这一方法的有效性为单细胞基因表达数据分析提供了新的视角。
🎯 应用场景
该研究的潜在应用领域包括生物医学研究、疾病诊断和个性化医疗等。通过提供一种简单且高效的分析方法,研究者可以更快速地处理和分析单细胞RNA测序数据,从而推动生物学和医学领域的进步。未来,该方法可能会影响单细胞基因组学的研究方向,促进更广泛的应用。
📄 摘要(原文)
Single-cell RNA sequencing (scRNA-seq) data exhibit strong and reproducible statistical structure. This has motivated the development of large-scale foundation models, such as TranscriptFormer, that use transformer-based architectures to learn a generative model for gene expression by embedding genes into a latent vector space. These embeddings have been used to obtain state-of-the-art (SOTA) performance on downstream tasks such as cell-type classification, disease-state prediction, and cross-species learning. Here, we ask whether similar performance can be achieved without utilizing computationally intensive deep learning-based representations. Using simple, interpretable pipelines that rely on careful normalization and linear methods, we obtain SOTA or near SOTA performance across multiple benchmarks commonly used to evaluate single-cell foundation models, including outperforming foundation models on out-of-distribution tasks involving novel cell types and organisms absent from the training data. Our findings highlight the need for rigorous benchmarking and suggest that the biology of cell identity can be captured by simple linear representations of single cell gene expression data.