Order Matters in Retrosynthesis: Structure-aware Generation via Reaction-Center-Guided Discrete Flow Matching
作者: Chenguang Wang, Zihan Zhou, Lei Bai, Tianshu Yu
分类: cs.LG
发布日期: 2026-02-13
💡 一句话要点
提出反应中心引导的离散流匹配方法RetroDiT,用于结构感知的逆合成生成。
🎯 匹配领域: 支柱二:RL算法与架构 (RL & Architecture) 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 逆合成分析 图神经网络 Transformer 离散流匹配 反应中心预测 分子生成
📋 核心要点
- 现有无模板逆合成方法学习效率低,半模板方法依赖于刚性反应库,泛化能力受限。
- 通过将反应中心原子置于序列头部,编码化学反应的两阶段性质,从而将隐式化学知识转化为显式位置模式。
- 提出的RetroDiT模型在USPTO-50k和USPTO-Full数据集上取得了SOTA性能,且训练效率更高。
📝 摘要(中文)
本文提出了一种结构感知的无模板逆合成框架,通过将化学反应的两阶段性质编码为位置归纳偏置来解决现有方法的局限性。该方法将反应中心原子置于序列头部,将隐式化学知识转化为模型易于捕获的显式位置模式。提出的RetroDiT骨干网络,是一个带有旋转位置嵌入的图Transformer,利用这种排序来优先考虑化学关键区域。结合离散流匹配,该方法将训练与采样解耦,并能在20-50步内生成,而之前的扩散方法需要500步。在预测反应中心的情况下,该方法在USPTO-50k(61.2% top-1)和大规模USPTO-Full(51.3% top-1)上均实现了最先进的性能。使用oracle中心时,性能分别达到71.1%和63.4%,超过了在100亿个反应上训练的基础模型,同时使用的数据量级更少。消融研究进一步表明,结构先验优于暴力扩展:一个具有适当排序的280K参数模型与一个没有排序的65M参数模型相匹配。
🔬 方法详解
问题定义:逆合成问题旨在给定目标分子,预测其可能的反应物。现有的无模板方法将其视为黑盒序列生成,忽略了化学反应的结构信息,导致学习效率低下。半模板方法虽然利用了反应模板,但依赖于预定义的刚性反应库,限制了泛化能力。因此,如何有效地利用化学结构信息,提高逆合成模型的性能和泛化能力是一个关键问题。
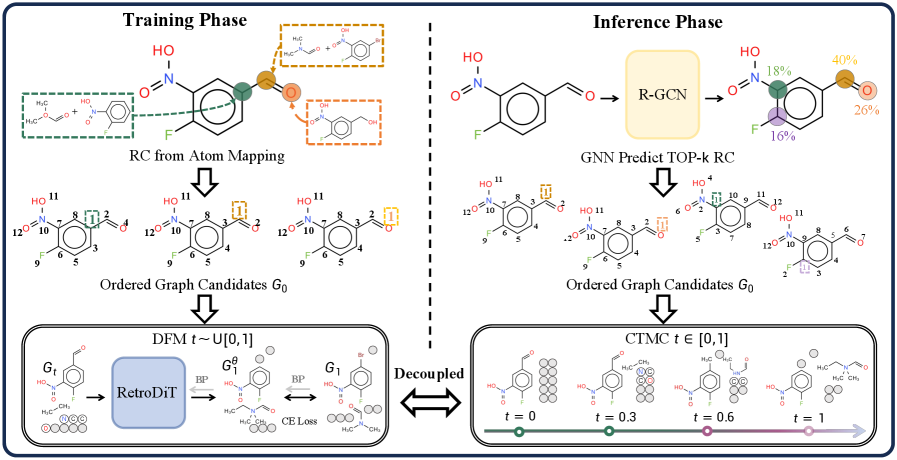
核心思路:论文的核心思路是利用原子排序来编码化学反应的结构信息。具体来说,将反应中心原子置于序列的头部,使得模型能够更容易地学习到反应中心与反应物之间的关系。这种排序方式将隐式的化学知识转化为显式的模型可学习的位置模式,从而提高了模型的学习效率和泛化能力。
技术框架:论文提出的RetroDiT框架主要包含两个部分:一是结构感知的编码器,用于将分子表示为序列;二是基于离散流匹配的生成器,用于生成反应物。编码器采用图Transformer结构,并引入旋转位置嵌入来编码原子之间的相对位置关系。生成器通过离散流匹配将训练与采样解耦,从而实现更快的生成速度。
关键创新:论文最重要的技术创新点在于利用原子排序来编码化学反应的结构信息。通过将反应中心原子置于序列头部,模型能够更容易地学习到反应中心与反应物之间的关系,从而提高了模型的性能和泛化能力。此外,论文还采用了离散流匹配方法,将训练与采样解耦,从而实现了更快的生成速度。
关键设计:RetroDiT使用图Transformer作为骨干网络,并引入旋转位置嵌入来编码原子之间的相对位置关系。损失函数采用标准的交叉熵损失函数。离散流匹配方法通过学习一个时间相关的向量场,将噪声分布映射到目标分布。采样过程通过求解一个常微分方程来实现,可以使用较少的步数生成高质量的分子。
🖼️ 关键图片
📊 实验亮点
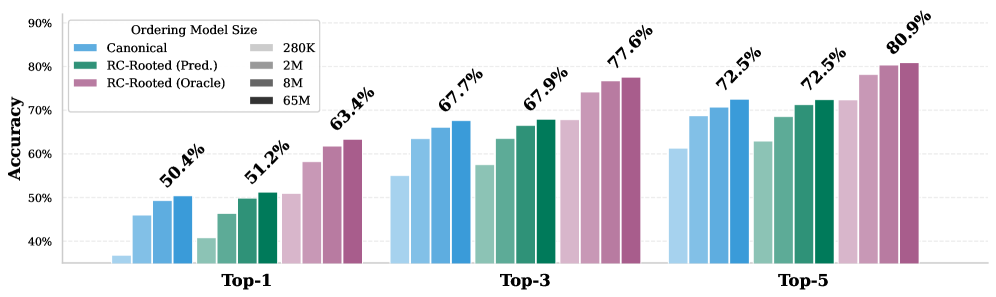
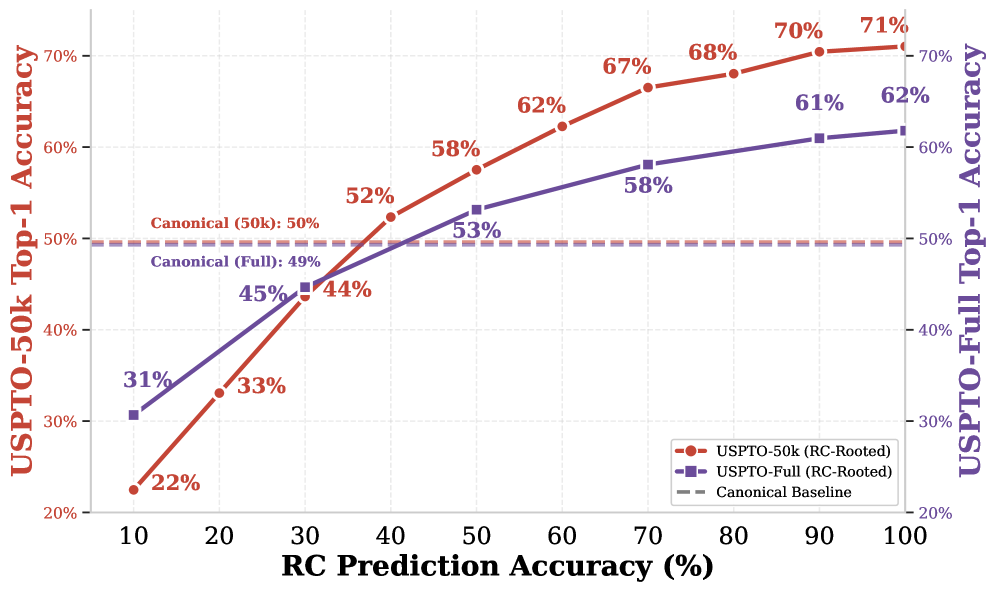
该方法在USPTO-50k数据集上取得了61.2%的top-1准确率,在USPTO-Full数据集上取得了51.3%的top-1准确率,均达到了SOTA水平。在oracle反应中心的情况下,性能分别达到71.1%和63.4%,超过了在100亿个反应上训练的基础模型,同时使用的数据量级更少。消融实验表明,结构先验优于暴力扩展,一个280K参数模型的效果可以匹配一个65M参数模型。
🎯 应用场景
该研究成果可应用于药物发现、材料科学等领域,加速新分子和新材料的研发过程。通过提高逆合成预测的准确性和效率,可以帮助化学家更快速地找到合成目标分子的可行路径,降低研发成本,缩短研发周期。未来,该方法有望与自动化合成平台结合,实现智能化化学合成。
📄 摘要(原文)
Template-free retrosynthesis methods treat the task as black-box sequence generation, limiting learning efficiency, while semi-template approaches rely on rigid reaction libraries that constrain generalization. We address this gap with a key insight: atom ordering in neural representations matters. Building on this insight, we propose a structure-aware template-free framework that encodes the two-stage nature of chemical reactions as a positional inductive bias. By placing reaction center atoms at the sequence head, our method transforms implicit chemical knowledge into explicit positional patterns that the model can readily capture. The proposed RetroDiT backbone, a graph transformer with rotary position embeddings, exploits this ordering to prioritize chemically critical regions. Combined with discrete flow matching, our approach decouples training from sampling and enables generation in 20--50 steps versus 500 for prior diffusion methods. Our method achieves state-of-the-art performance on both USPTO-50k (61.2% top-1) and the large-scale USPTO-Full (51.3% top-1) with predicted reaction centers. With oracle centers, performance reaches 71.1% and 63.4% respectively, surpassing foundation models trained on 10 billion reactions while using orders of magnitude less data. Ablation studies further reveal that structural priors outperform brute-force scaling: a 280K-parameter model with proper ordering matches a 65M-parameter model without it.