Modular Multi-Task Learning for Chemical Reaction Prediction
作者: Jiayun Pang, Ahmed M. Zaitoun, Xacobe Couso Cambeiro, Ivan Vulić
分类: cs.LG, cs.AI, cs.CL
发布日期: 2026-02-11
备注: 19 pages, 7 figures
💡 一句话要点
LoRA助力化学反应预测,实现参数高效的多任务学习
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 化学反应预测 大型语言模型 低秩适应 参数高效微调 多任务学习
📋 核心要点
- 现有方法难以在有限的化学反应数据集上微调大型语言模型,同时保持其通用化学知识。
- 论文提出使用低秩适应(LoRA)进行参数高效的微调,以在学习新反应知识的同时,保留通用化学理解。
- 实验表明,LoRA在反应预测、逆合成和试剂预测方面,性能与完全微调相当,且能有效缓解灾难性遗忘。
📝 摘要(中文)
在化学和药物研发中,将大型语言模型(LLM)在广泛的有机化学知识上训练后,应用于更小、特定领域的反应数据集是一个关键挑战。有效的专业化需要在学习新的反应知识的同时,保留跨相关任务的通用化学理解。本文评估了低秩适应(LoRA)作为一种参数高效的替代方案,以替代完全微调,用于在有限的复杂数据集上进行有机反应预测。使用USPTO反应类别和具有挑战性的C-H官能化反应,我们对正向反应预测、逆合成和试剂预测进行了基准测试。LoRA实现了与完全微调相当的准确性,同时有效地减轻了灾难性遗忘,并更好地保持了多任务性能。两种微调方法都泛化到训练分布之外,产生合理的替代溶剂预测。值得注意的是,C-H官能化微调表明,LoRA和完全微调编码了略有不同的反应模式,表明LoRA可以更有效地进行反应特异性适应。随着LLM的持续扩展,我们的结果突出了模块化、参数高效的微调策略在化学应用中的灵活部署的实用性。
🔬 方法详解
问题定义:论文旨在解决将大型语言模型(LLM)应用于特定化学反应预测任务时,在数据量有限的情况下,如何有效微调模型,使其既能学习新的反应知识,又能保留其通用的化学理解能力的问题。现有方法,如完全微调,容易导致灾难性遗忘,并且参数效率较低,难以适应不断扩展的LLM。
核心思路:论文的核心思路是利用低秩适应(LoRA)作为一种参数高效的微调方法。LoRA通过在预训练模型的权重矩阵旁添加低秩矩阵,只训练这些低秩矩阵,从而大大减少了需要训练的参数量,降低了过拟合的风险,并能更好地保留预训练模型的通用知识。
技术框架:整体框架包括以下步骤:1) 选择一个预训练的LLM;2) 在LLM的权重矩阵旁添加低秩矩阵;3) 使用特定的化学反应数据集(如USPTO反应类别或C-H官能化反应)对低秩矩阵进行微调;4) 评估微调后的模型在正向反应预测、逆合成和试剂预测等任务上的性能。
关键创新:最重要的技术创新点在于将LoRA应用于化学反应预测任务,并证明了其在参数效率、防止灾难性遗忘和保持多任务性能方面的优势。与完全微调相比,LoRA能够以更少的参数实现相当甚至更好的性能,并且能够编码略有不同的反应模式,从而实现更有效的反应特异性适应。
关键设计:论文的关键设计包括:1) 选择合适的低秩矩阵的秩(rank);2) 使用特定的损失函数来优化低秩矩阵,例如交叉熵损失;3) 对比LoRA和完全微调在不同任务上的性能,并分析其编码的反应模式的差异。
🖼️ 关键图片

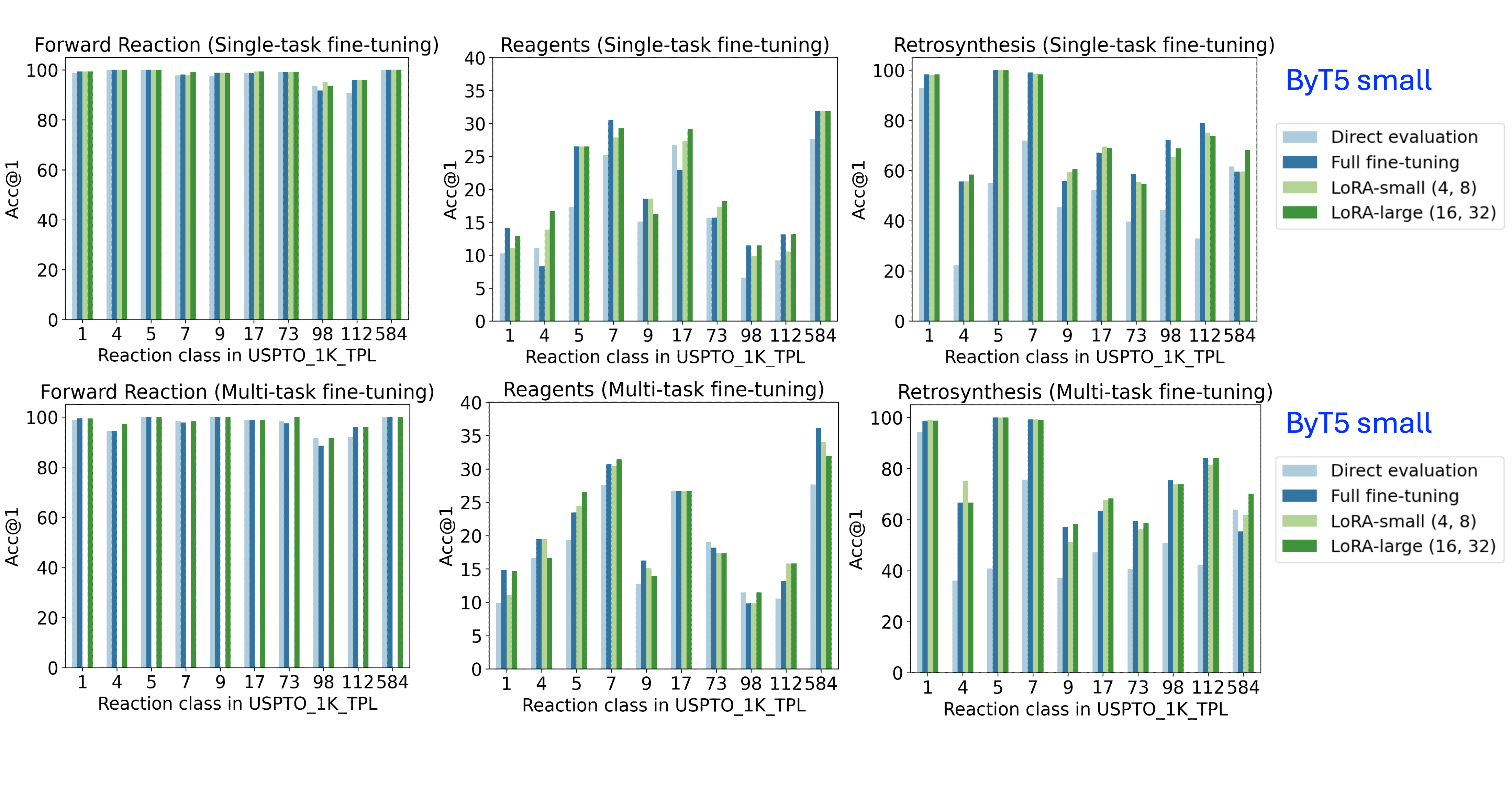
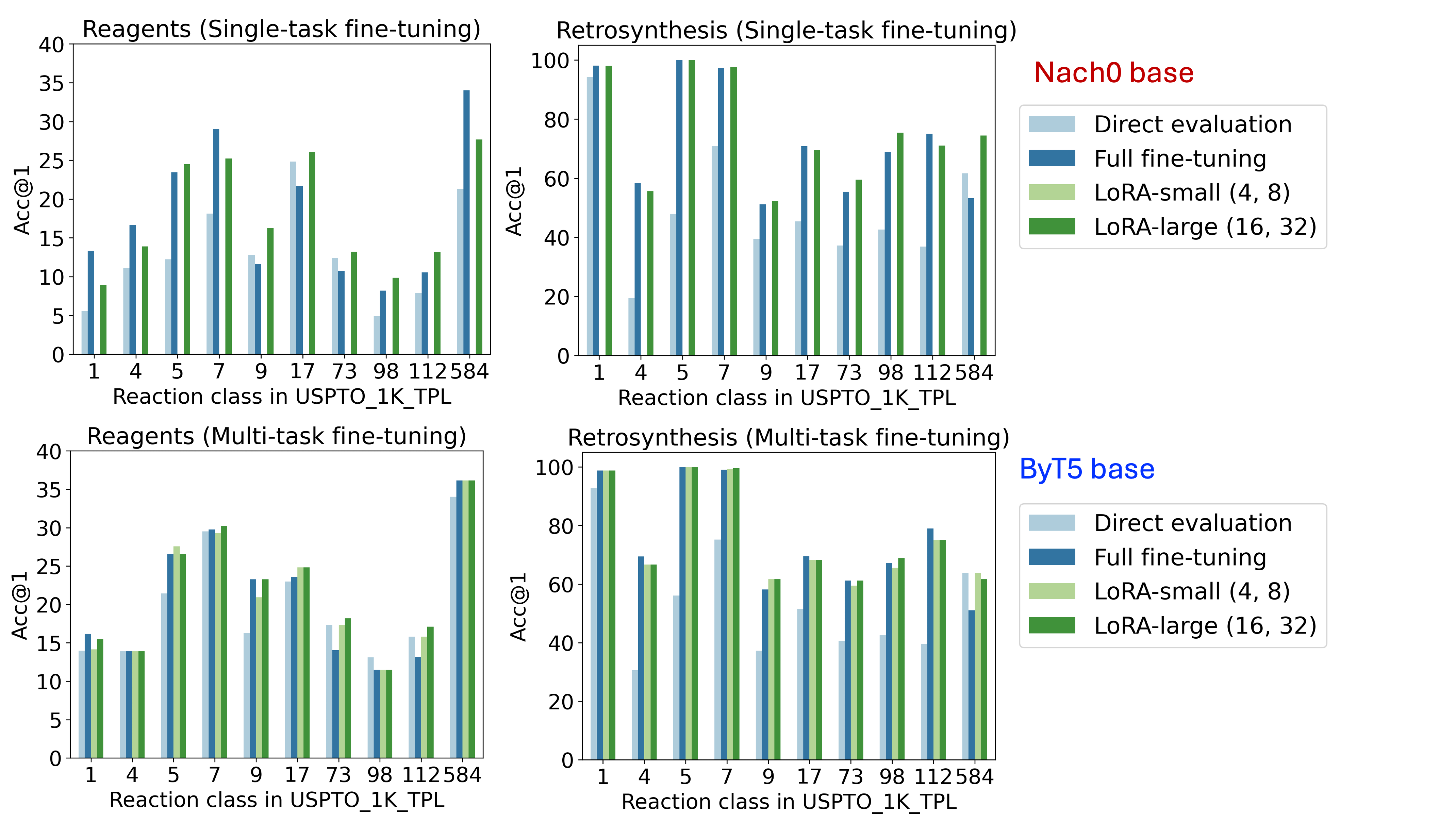
📊 实验亮点
实验结果表明,LoRA在USPTO反应类别和C-H官能化反应数据集上,实现了与完全微调相当的准确性,同时有效地减轻了灾难性遗忘,并更好地保持了多任务性能。特别是在C-H官能化微调中,LoRA和完全微调编码了略有不同的反应模式,表明LoRA可以更有效地进行反应特异性适应。此外,两种微调方法都泛化到训练分布之外,产生合理的替代溶剂预测。
🎯 应用场景
该研究成果可应用于化学和药物研发领域,加速新反应的发现和优化,降低实验成本。通过参数高效的微调,可以更灵活地将大型语言模型应用于各种化学任务,例如反应预测、逆合成、试剂推荐和溶剂选择等,从而提高研发效率和成功率。未来,该方法有望扩展到其他科学领域,例如材料科学和生物学。
📄 摘要(原文)
Adapting large language models (LLMs) trained on broad organic chemistry to smaller, domain-specific reaction datasets is a key challenge in chemical and pharmaceutical R&D. Effective specialisation requires learning new reaction knowledge while preserving general chemical understanding across related tasks. Here, we evaluate Low-Rank Adaptation (LoRA) as a parameter-efficient alternative to full fine-tuning for organic reaction prediction on limited, complex datasets. Using USPTO reaction classes and challenging C-H functionalisation reactions, we benchmark forward reaction prediction, retrosynthesis and reagent prediction. LoRA achieves accuracy comparable to full fine-tuning while effectively mitigating catastrophic forgetting and better preserving multi-task performance. Both fine-tuning approaches generalise beyond training distributions, producing plausible alternative solvent predictions. Notably, C-H functionalisation fine-tuning reveals that LoRA and full fine-tuning encode subtly different reactivity patterns, suggesting more effective reaction-specific adaptation with LoRA. As LLMs continue to scale, our results highlight the practicality of modular, parameter-efficient fine-tuning strategies for their flexible deployment for chemistry applications.