TerraBind: Fast and Accurate Binding Affinity Prediction through Coarse Structural Representations
作者: Matteo Rossi, Ryan Pederson, Miles Wang-Henderson, Ben Kaufman, Edward C. Williams, Carl Underkoffler, Owen Lewis Howell, Adrian Layer, Stephan Thaler, Narbe Mardirossian, John Anthony Parkhill
分类: cs.LG, q-bio.BM
发布日期: 2026-02-08
备注: 31 pages, 14 figures
💡 一句话要点
TerraBind:通过粗粒度结构表征实现快速准确的蛋白-配体结合亲和力预测
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 药物发现 结合亲和力预测 蛋白质-配体相互作用 粗粒度建模 多模态学习
📋 核心要点
- 现有基于结构的药物设计方法依赖于计算成本高的全原子扩散,导致推理速度慢,难以进行大规模化合物筛选。
- TerraBind采用粗粒度的口袋级别表示,结合COATI-3分子编码和ESM-2蛋白质嵌入,学习高效的结构表示,用于快速姿势生成和亲和力预测。
- 实验表明,TerraBind在保持姿势预测准确性的同时,显著提升了结合亲和力预测的准确性和速度,并在模拟药物发现周期中表现出优异的性能。
📝 摘要(中文)
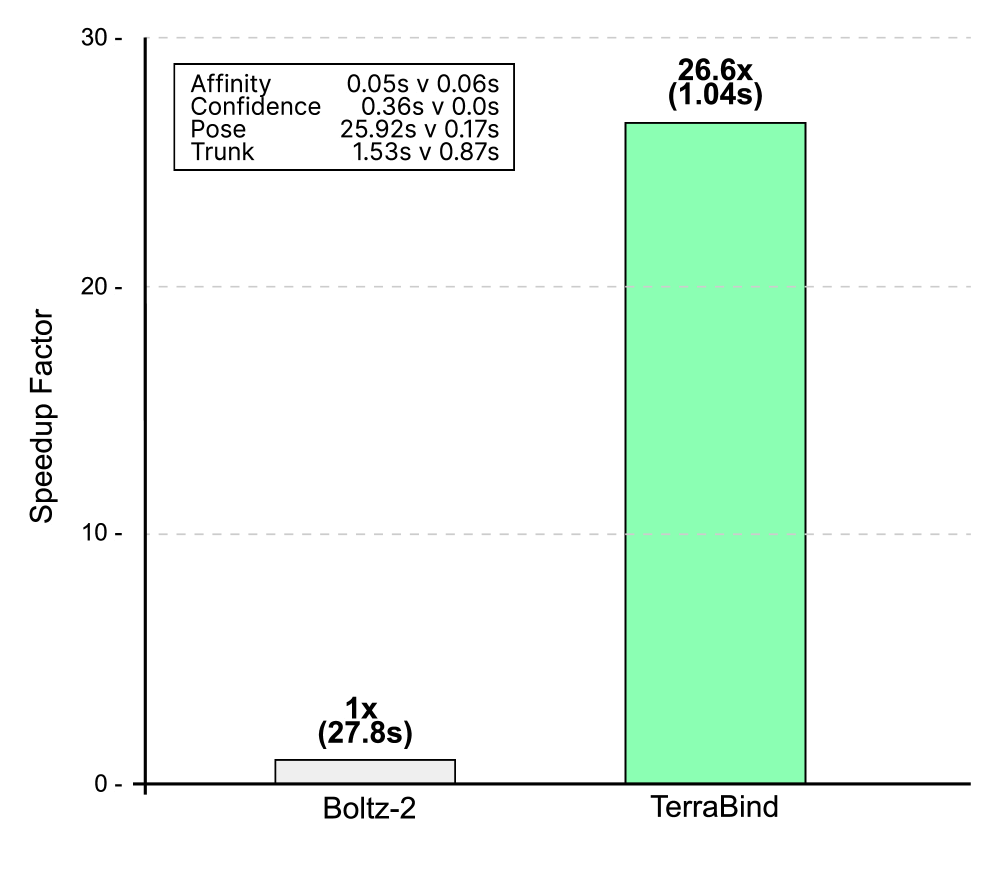
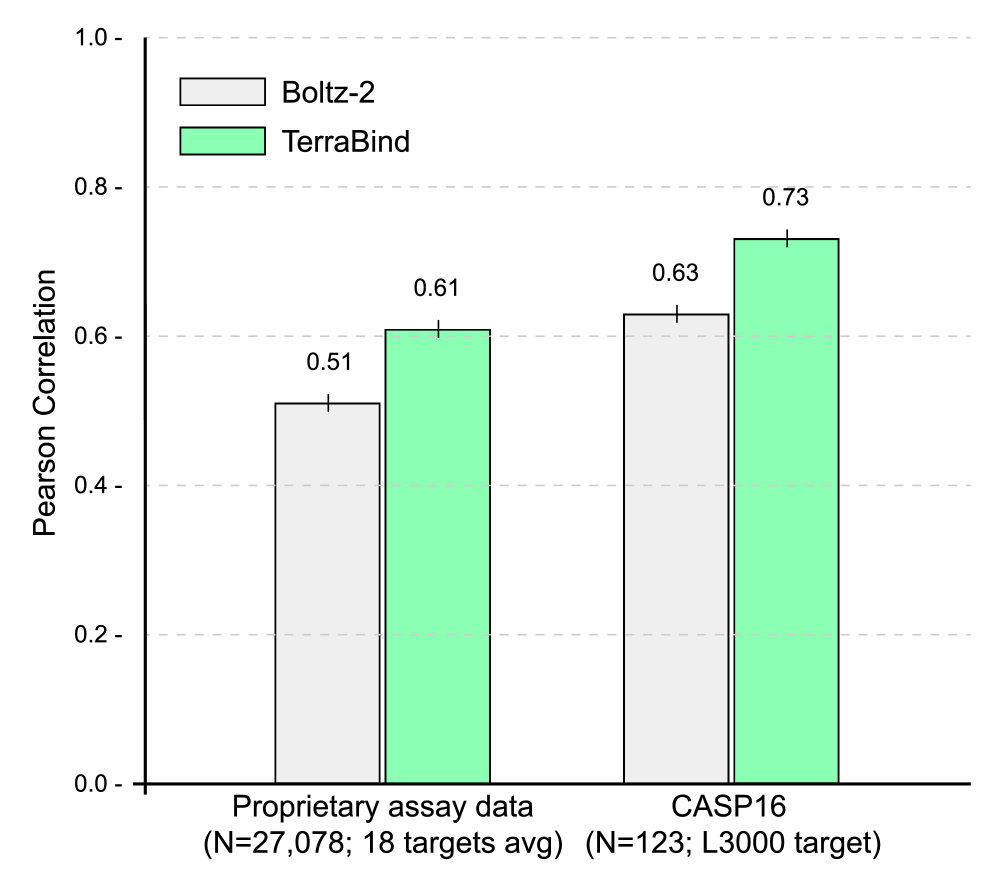
TerraBind是一个用于蛋白质-配体结构和结合亲和力预测的基础模型,其推理速度比最先进的方法快26倍,同时将亲和力预测准确度提高了约20%。目前基于结构的药物设计的深度学习方法依赖于昂贵的全原子扩散来生成3D坐标,这造成了推理瓶颈,使得大规模化合物筛选在计算上难以实现。我们用一个关键假设挑战了这种范式:对于准确的小分子姿势和结合亲和力预测,完全的全原子分辨率是不必要的。TerraBind通过粗粒度的口袋级别表示(仅蛋白质Cβ原子和配体重原子)在一个多模态架构中测试了这个假设,该架构结合了COATI-3分子编码和ESM-2蛋白质嵌入,学习丰富的结构表示,这些表示用于无扩散优化模块进行姿势生成和结合亲和力似然预测模块。在结构预测基准测试(FoldBench、PoseBusters、Runs N' Poses)中,TerraBind在配体姿势准确性方面与基于扩散的基线相匹配。关键的是,在公共基准(CASP16)和多样化的专有数据集(18个生化/细胞分析)上,TerraBind在结合亲和力预测的Pearson相关性方面优于Boltz-2约20%。我们表明,亲和力预测模块还提供了良好校准的亲和力不确定性估计,解决了药物发现中可靠化合物优先级排序的关键差距。此外,该模块支持持续学习框架和对冲批量选择策略,在模拟药物发现周期中,与基于贪婪的方法相比,所选分子的亲和力提高了6倍。
🔬 方法详解
问题定义:论文旨在解决基于结构的药物设计中,现有方法依赖全原子扩散导致计算成本高、推理速度慢的问题。现有方法难以进行大规模化合物筛选,限制了药物发现的效率。
核心思路:论文的核心思路是,对于准确的配体姿势和结合亲和力预测,并不需要完全的全原子分辨率。通过使用粗粒度的口袋级别表示,可以在显著降低计算复杂度的同时,保持甚至提高预测的准确性。
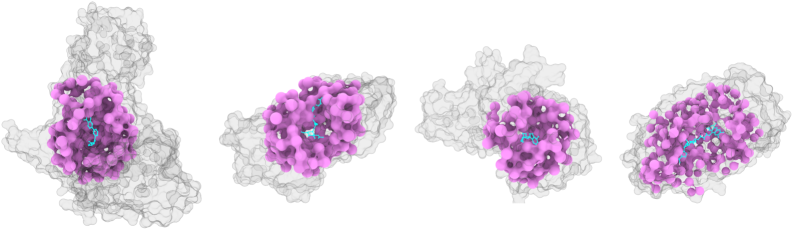
技术框架:TerraBind采用多模态架构,包含以下主要模块:1) 粗粒度结构表示:仅使用蛋白质Cβ原子和配体重原子。2) 分子编码:使用COATI-3对配体进行编码。3) 蛋白质嵌入:使用ESM-2对蛋白质进行嵌入。4) 无扩散优化模块:用于配体姿势生成。5) 结合亲和力似然预测模块:用于预测结合亲和力,并提供不确定性估计。
关键创新:最重要的技术创新点在于使用粗粒度的结构表示,避免了昂贵的全原子扩散过程,从而显著提高了推理速度。此外,结合COATI-3和ESM-2的多模态架构,以及用于亲和力预测的不确定性估计,也都是重要的创新点。
关键设计:TerraBind的关键设计包括:1) 使用Cβ原子和配体重原子进行粗粒度表示,平衡了计算效率和信息保留。2) COATI-3和ESM-2的选择,保证了分子和蛋白质编码的质量。3) 无扩散优化模块的设计,实现了快速的配体姿势生成。4) 亲和力预测模块的设计,不仅预测亲和力,还提供了校准良好的不确定性估计。
🖼️ 关键图片
📊 实验亮点
TerraBind在推理速度上比最先进的方法快26倍,同时在结合亲和力预测的Pearson相关性方面,在CASP16和专有数据集上均优于Boltz-2约20%。在结构预测基准测试中,TerraBind在配体姿势准确性方面与基于扩散的基线相匹配。在模拟药物发现周期中,TerraBind的持续学习框架和对冲批量选择策略,使所选分子的亲和力提高了6倍(相比于贪婪方法)。
🎯 应用场景
TerraBind可应用于药物发现领域,加速先导化合物的筛选和优化。其快速的推理速度和准确的亲和力预测能力,能够显著提高药物研发的效率,降低研发成本。此外,TerraBind提供的亲和力不确定性估计,有助于更可靠地进行化合物优先级排序,提高药物研发的成功率。
📄 摘要(原文)
We present TerraBind, a foundation model for protein-ligand structure and binding affinity prediction that achieves 26-fold faster inference than state-of-the-art methods while improving affinity prediction accuracy by $\sim$20\%. Current deep learning approaches to structure-based drug design rely on expensive all-atom diffusion to generate 3D coordinates, creating inference bottlenecks that render large-scale compound screening computationally intractable. We challenge this paradigm with a critical hypothesis: full all-atom resolution is unnecessary for accurate small molecule pose and binding affinity prediction. TerraBind tests this hypothesis through a coarse pocket-level representation (protein C$_β$ atoms and ligand heavy atoms only) within a multimodal architecture combining COATI-3 molecular encodings and ESM-2 protein embeddings that learns rich structural representations, which are used in a diffusion-free optimization module for pose generation and a binding affinity likelihood prediction module. On structure prediction benchmarks (FoldBench, PoseBusters, Runs N' Poses), TerraBind matches diffusion-based baselines in ligand pose accuracy. Crucially, TerraBind outperforms Boltz-2 by $\sim$20\% in Pearson correlation for binding affinity prediction on both a public benchmark (CASP16) and a diverse proprietary dataset (18 biochemical/cell assays). We show that the affinity prediction module also provides well-calibrated affinity uncertainty estimates, addressing a critical gap in reliable compound prioritization for drug discovery. Furthermore, this module enables a continual learning framework and a hedged batch selection strategy that, in simulated drug discovery cycles, achieves 6$\times$ greater affinity improvement of selected molecules over greedy-based approaches.