Interpretable Perturbation Modeling Through Biomedical Knowledge Graphs
作者: Pascal Passigan, Kevin Zhu, Angelina Ning
分类: cs.LG, cs.AI
发布日期: 2025-12-24 (更新: 2025-12-31)
💡 一句话要点
提出基于生物医学知识图谱的可解释扰动建模框架,用于预测药物对基因表达的影响。
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 生物医学知识图谱 图神经网络 基因扰动预测 药物发现 转录组学
📋 核心要点
- 现有方法在药物作用机制理解上,主要集中于二元药物-疾病关联,缺乏对基因扰动等转录组效应的深入分析。
- 论文构建融合多源信息的生物医学知识图谱,并利用图注意力网络学习药物-细胞对的基因表达变化。
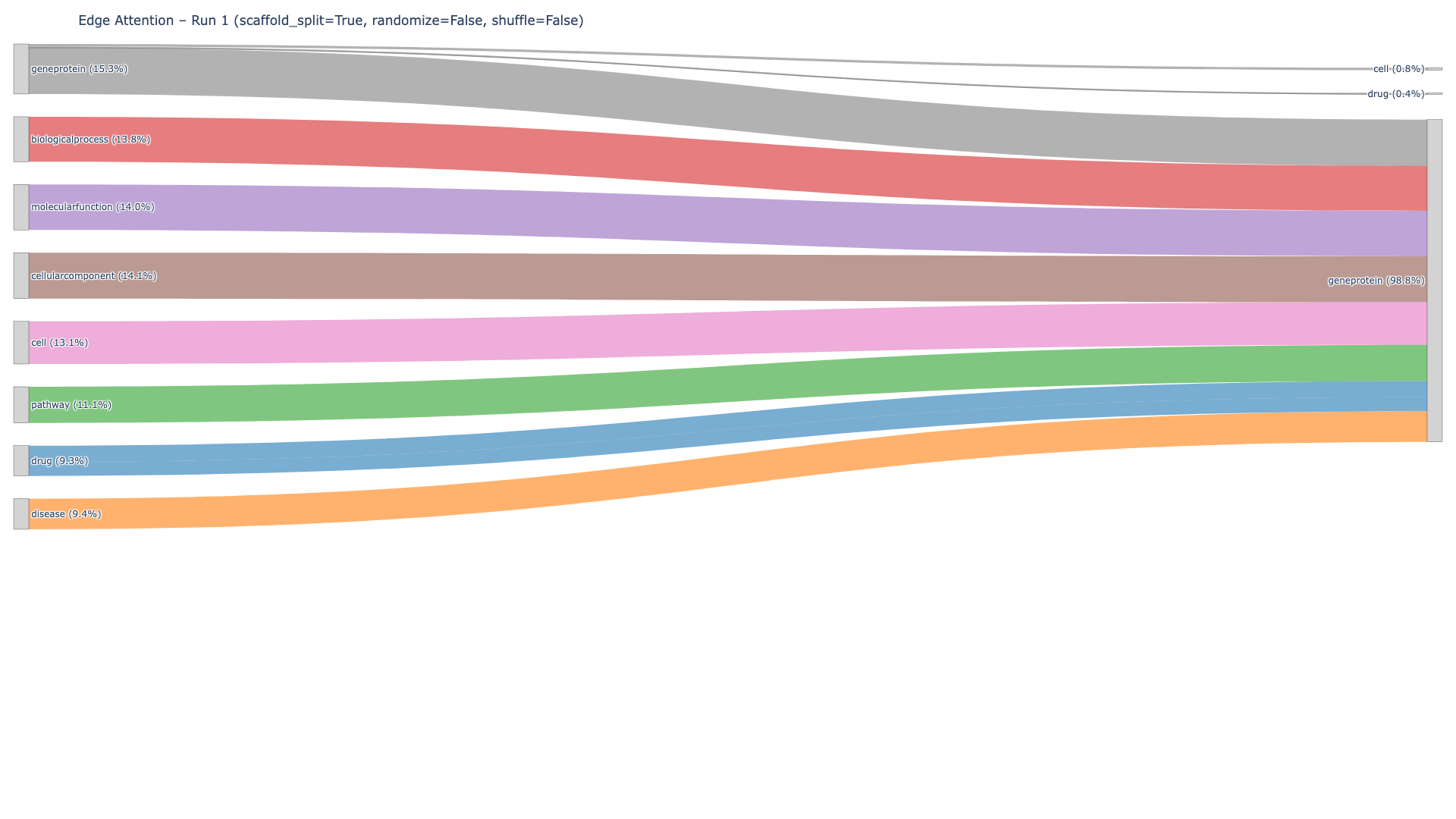
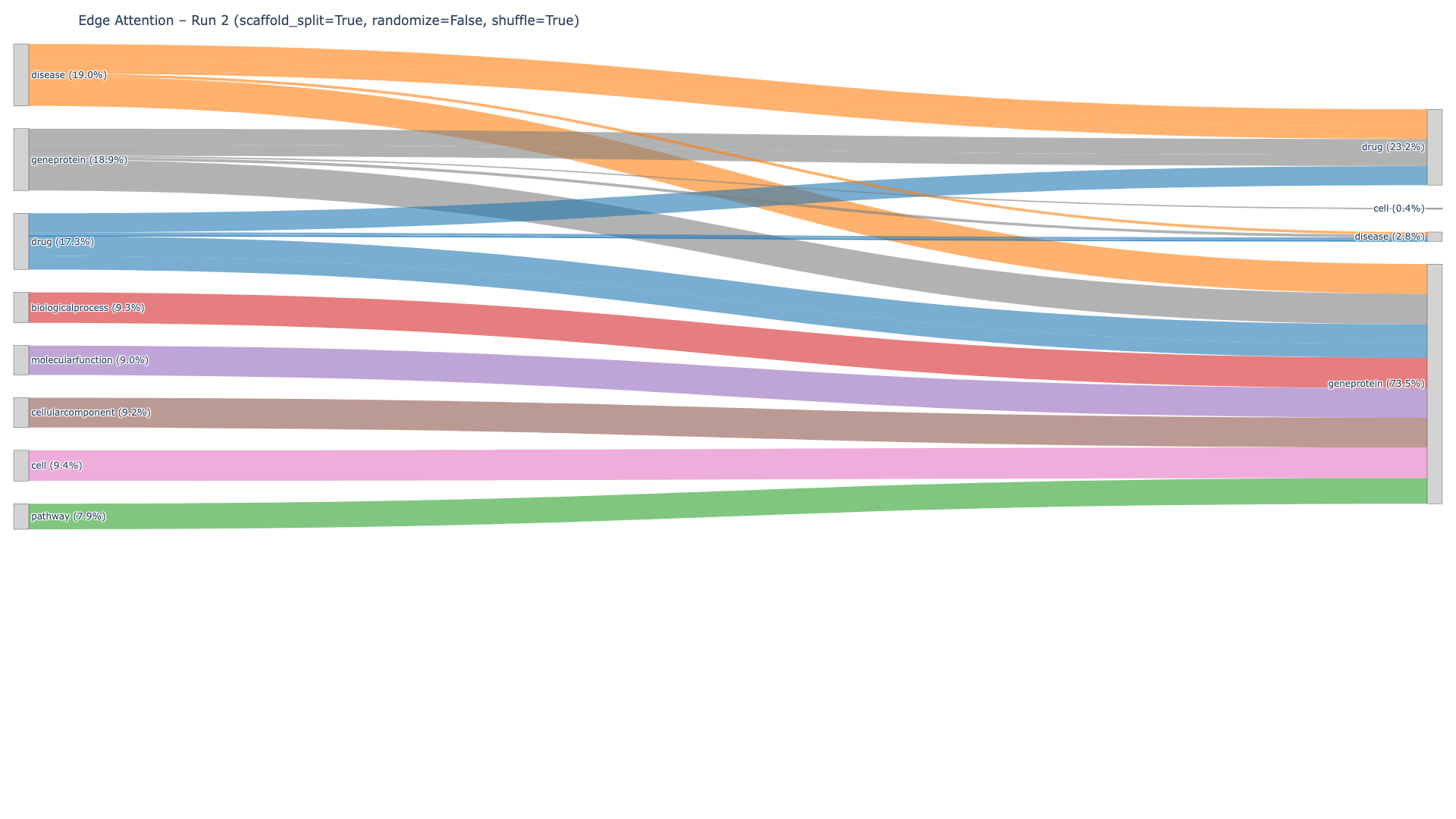
- 实验结果表明,该框架在预测差异表达基因方面优于传统方法,并验证了知识图谱边缘信息的重要性。
📝 摘要(中文)
理解小分子如何扰乱基因表达对于揭示药物机制、预测脱靶效应和识别再利用机会至关重要。虽然先前的深度学习框架已将多模态嵌入集成到生物医学知识图谱(BKG)中,并通过图神经网络消息传递范式进一步改进了这些表示,但这些模型主要应用于链接预测和二元药物-疾病关联等任务,而非基因扰动任务,而基因扰动可能揭示更多关于机制性转录组效应的信息。为了解决这一差距,我们构建了一个合并的生物医学图,该图集成了(i) PrimeKG++,它是PrimeKG的增强版本,包含节点语义丰富的嵌入,以及(ii) LINCS L1000药物和细胞系节点,这些节点使用来自MolFormerXL和BioBERT等基础模型的多模态嵌入进行初始化。使用这个异构图,我们训练了一个图注意力网络(GAT),它带有一个下游预测头,用于学习给定药物-细胞对的978个标志基因的delta表达谱。结果表明,我们的框架在差异表达基因(DEG)方面优于MLP基线——MLP基线预测药物特征、靶标特征和基线细胞表达的连接嵌入给出的delta表达——在scaffold和随机分割下均表现良好。边缘洗牌和节点特征随机化的消融实验进一步表明,生物医学知识图谱提供的边缘增强了扰动水平的预测。更广泛地说,我们的框架为机制性药物建模提供了一条路径:从二元药物-疾病关联任务转向治疗干预的精细转录效应。
🔬 方法详解
问题定义:论文旨在解决药物对基因表达的扰动预测问题,现有方法主要关注药物-疾病关联,忽略了药物对基因表达的精细影响。现有方法的痛点在于无法有效利用多模态生物医学数据,难以准确预测药物的转录组效应。
核心思路:论文的核心思路是构建一个融合多源信息的生物医学知识图谱,并利用图神经网络学习药物、靶标和细胞之间的复杂关系,从而预测药物对基因表达的扰动。这种方法能够有效整合多模态数据,并捕捉药物作用的潜在机制。
技术框架:整体框架包含以下几个主要步骤:1) 构建融合PrimeKG++和LINCS L1000数据的生物医学知识图谱;2) 使用MolFormerXL和BioBERT等预训练模型初始化节点嵌入;3) 使用图注意力网络(GAT)学习节点表示;4) 使用下游预测头预测药物-细胞对的基因表达变化。
关键创新:论文的关键创新在于将生物医学知识图谱与图神经网络相结合,用于预测药物对基因表达的扰动。与传统方法相比,该方法能够更有效地利用多模态生物医学数据,并捕捉药物作用的潜在机制。此外,论文还通过消融实验验证了知识图谱边缘信息的重要性。
关键设计:论文使用图注意力网络(GAT)作为主要的图神经网络模型,GAT能够根据邻居节点的重要性动态调整权重,从而更好地学习节点表示。损失函数采用均方误差(MSE),用于衡量预测的基因表达变化与真实值之间的差异。节点嵌入的初始化使用了MolFormerXL和BioBERT等预训练模型,这些模型能够提供丰富的语义信息。
🖼️ 关键图片

📊 实验亮点
实验结果表明,该框架在预测差异表达基因(DEG)方面优于MLP基线,在scaffold和随机分割下均表现良好。边缘洗牌和节点特征随机化的消融实验进一步验证了生物医学知识图谱提供的边缘信息对扰动水平预测的增强作用。这些结果表明,该框架能够有效利用生物医学知识图谱,提高药物扰动预测的准确性。
🎯 应用场景
该研究成果可应用于药物发现、药物重定位和个性化医疗等领域。通过预测药物对基因表达的影响,可以更准确地评估药物的疗效和安全性,从而加速药物研发进程。此外,该方法还可以用于识别潜在的药物重定位机会,并为患者提供更个性化的治疗方案。
📄 摘要(原文)
Understanding how small molecules perturb gene expression is essential for uncovering drug mechanisms, predicting off-target effects, and identifying repurposing opportunities. While prior deep learning frameworks have integrated multimodal embeddings into biomedical knowledge graphs (BKGs) and further improved these representations through graph neural network message-passing paradigms, these models have been applied to tasks such as link prediction and binary drug-disease association, rather than the task of gene perturbation, which may unveil more about mechanistic transcriptomic effects. To address this gap, we construct a merged biomedical graph that integrates (i) PrimeKG++, an augmentation of PrimeKG containing semantically rich embeddings for nodes with (ii) LINCS L1000 drug and cell line nodes, initialized with multimodal embeddings from foundation models such as MolFormerXL and BioBERT. Using this heterogeneous graph, we train a graph attention network (GAT) with a downstream prediction head that learns the delta expression profile of over 978 landmark genes for a given drug-cell pair. Our results show that our framework outperforms MLP baselines for differentially expressed genes (DEG) -- which predict the delta expression given a concatenated embedding of drug features, target features, and baseline cell expression -- under the scaffold and random splits. Ablation experiments with edge shuffling and node feature randomization further demonstrate that the edges provided by biomedical KGs enhance perturbation-level prediction. More broadly, our framework provides a path toward mechanistic drug modeling: moving beyond binary drug-disease association tasks to granular transcriptional effects of therapeutic intervention.