When Genes Speak: A Semantic-Guided Framework for Spatially Resolved Transcriptomics Data Clustering
作者: Jiangkai Long, Yanran Zhu, Chang Tang, Kun Sun, Yuanyuan Liu, Xuesong Yan
分类: cs.LG
发布日期: 2025-11-14
备注: AAAI'2026 poster paper. 12 pages, 8 figures
💡 一句话要点
SemST:提出一种语义引导的深度学习框架,用于空间转录组数据聚类。
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 空间转录组学 数据聚类 深度学习 图神经网络 语义嵌入
📋 核心要点
- 现有空间转录组学方法忽略了基因符号中蕴含的丰富生物学语义,限制了对组织微环境的深入理解。
- SemST利用大型语言模型将基因集转化为具有生物学意义的嵌入,并与空间邻域关系融合,实现生物学功能和空间结构的整合。
- 实验表明,SemST在聚类性能上达到了最先进水平,并且FSM模块具有通用性,能提升其他基线方法的性能。
📝 摘要(中文)
空间转录组学能够在空间背景下进行基因表达谱分析,为组织微环境提供前所未有的洞察。然而,大多数计算模型将基因视为孤立的数值特征,忽略了其符号中蕴含的丰富生物学语义,阻碍了对关键生物学特征的深入理解。为了克服这一局限,我们提出了SemST,一种语义引导的深度学习框架,用于空间转录组数据聚类。SemST利用大型语言模型(LLMs)使基因能够通过其符号意义“说话”,将每个组织位点内的基因集转化为具有生物学意义的嵌入。然后,将这些嵌入与图神经网络(GNNs)捕获的空间邻域关系融合,实现生物学功能和空间结构的连贯整合。我们进一步引入了细粒度语义调制(FSM)模块,以最佳地利用这些生物学先验知识。FSM模块学习特定于位点的仿射变换,使语义嵌入能够对空间特征进行元素级校准,从而将高阶生物学知识动态地注入到空间背景中。在公共空间转录组数据集上的大量实验表明,SemST实现了最先进的聚类性能。至关重要的是,FSM模块具有即插即用的通用性,在集成到其他基线方法中时,始终如一地提高了性能。
🔬 方法详解
问题定义:空间转录组学数据聚类的目标是将组织样本中的不同细胞类型或状态区分开来。现有方法主要将基因表达数据视为数值特征,忽略了基因名称本身所蕴含的生物学信息,导致聚类结果缺乏生物学解释性。
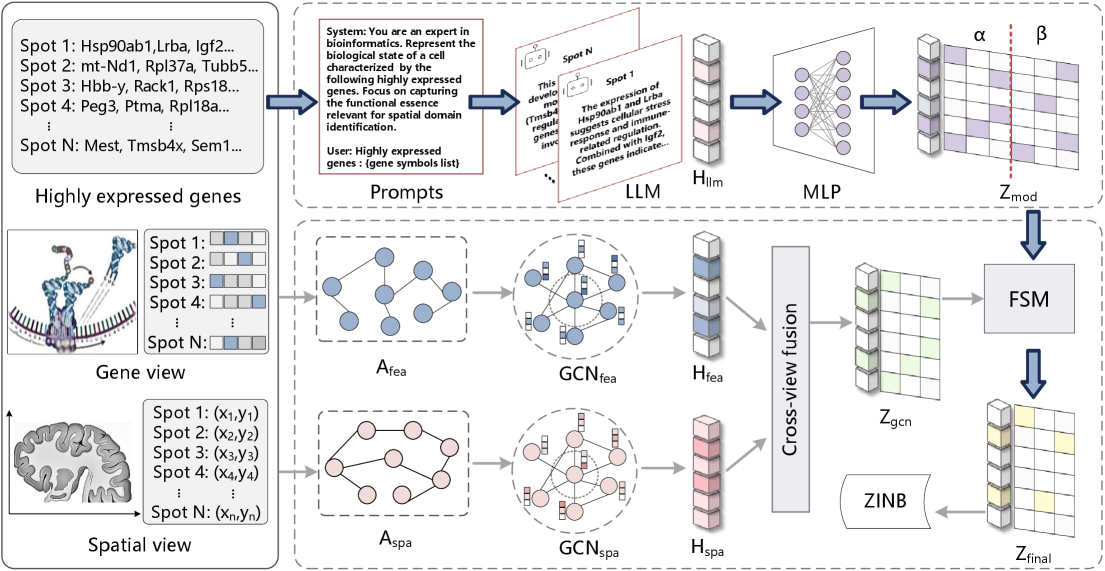
核心思路:SemST的核心思路是利用大型语言模型(LLMs)将基因名称转化为语义嵌入,从而将基因的生物学信息融入到聚类过程中。通过将语义嵌入与空间信息相结合,SemST能够更准确地识别组织样本中的不同细胞类型或状态。
技术框架:SemST的整体框架包括三个主要模块:1) 基因语义嵌入模块:利用LLMs将每个组织位点内的基因集转化为生物学意义的嵌入。2) 空间信息提取模块:使用图神经网络(GNNs)捕获空间邻域关系。3) 细粒度语义调制(FSM)模块:学习特定于位点的仿射变换,使语义嵌入能够对空间特征进行元素级校准,从而将高阶生物学知识动态地注入到空间背景中。最终,将语义嵌入和空间信息融合后进行聚类。
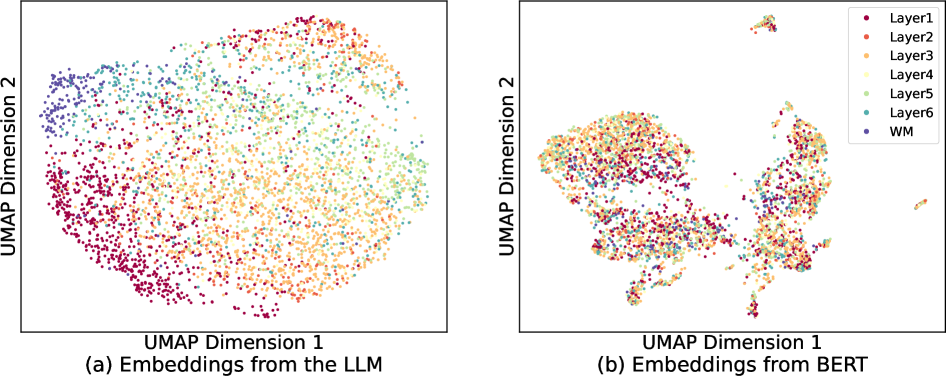
关键创新:SemST的关键创新在于利用LLMs将基因的生物学信息融入到空间转录组数据聚类中。传统的聚类方法只考虑基因表达的数值特征,而SemST通过引入基因的语义信息,能够更准确地识别组织样本中的不同细胞类型或状态。此外,FSM模块的设计也使得语义信息能够更好地与空间信息融合。
关键设计:SemST使用预训练的LLMs(具体模型未知)来生成基因的语义嵌入。GNN的具体结构也未知,但其目标是捕获空间邻域关系。FSM模块学习的仿射变换的具体参数设置也未知,但其目标是使语义嵌入能够对空间特征进行元素级校准。损失函数的设计也未知,但其目标是优化聚类结果,并使语义信息和空间信息更好地融合。
🖼️ 关键图片

📊 实验亮点
SemST在多个公共空间转录组数据集上取得了最先进的聚类性能。FSM模块具有即插即用的特性,可以显著提升其他基线方法的性能。具体性能提升数据未知,但论文强调了SemST和FSM模块的有效性和通用性。
🎯 应用场景
SemST可应用于多种生物医学研究领域,例如肿瘤微环境研究、发育生物学研究和疾病诊断等。通过更准确地识别组织样本中的不同细胞类型或状态,SemST能够帮助研究人员深入了解疾病的发生发展机制,并为开发新的治疗方法提供线索。此外,SemST还可以用于药物靶点发现和生物标志物识别。
📄 摘要(原文)
Spatial transcriptomics enables gene expression profiling with spatial context, offering unprecedented insights into the tissue microenvironment. However, most computational models treat genes as isolated numerical features, ignoring the rich biological semantics encoded in their symbols. This prevents a truly deep understanding of critical biological characteristics. To overcome this limitation, we present SemST, a semantic-guided deep learning framework for spatial transcriptomics data clustering. SemST leverages Large Language Models (LLMs) to enable genes to "speak" through their symbolic meanings, transforming gene sets within each tissue spot into biologically informed embeddings. These embeddings are then fused with the spatial neighborhood relationships captured by Graph Neural Networks (GNNs), achieving a coherent integration of biological function and spatial structure. We further introduce the Fine-grained Semantic Modulation (FSM) module to optimally exploit these biological priors. The FSM module learns spot-specific affine transformations that empower the semantic embeddings to perform an element-wise calibration of the spatial features, thus dynamically injecting high-order biological knowledge into the spatial context. Extensive experiments on public spatial transcriptomics datasets show that SemST achieves state-of-the-art clustering performance. Crucially, the FSM module exhibits plug-and-play versatility, consistently improving the performance when integrated into other baseline methods.