Pearl: A Foundation Model for Placing Every Atom in the Right Location
作者: Genesis Research Team, Alejandro Dobles, Nina Jovic, Kenneth Leidal, Pranav Murugan, David C. Williams, Drausin Wulsin, Nate Gruver, Christina X. Ji, Korrawat Pruegsanusak, Gianluca Scarpellini, Ansh Sharma, Wojciech Swiderski, Andrea Bootsma, Richard Strong Bowen, Charlotte Chen, Jamin Chen, Marc André Dämgen, Benjamin DiFrancesco, J. D. Fishman, Alla Ivanova, Zach Kagin, David Li-Bland, Zuli Liu, Igor Morozov, Jeffrey Ouyang-Zhang, Frank C. Pickard, Kushal S. Shah, Ben Shor, Gabriel Monteiro da Silva, Roy Tal, Maxx Tessmer, Carl Tilbury, Cyr Vetcher, Daniel Zeng, Maruan Al-Shedivat, Aleksandra Faust, Evan N. Feinberg, Michael V. LeVine, Matteus Pan
分类: cs.LG, q-bio.QM
发布日期: 2025-10-28 (更新: 2025-10-29)
备注: technical report
💡 一句话要点
Pearl:用于蛋白质-配体共折叠的原子级精度基础模型
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 蛋白质-配体共折叠 药物发现 深度学习 扩散模型 SO(3)等变性
📋 核心要点
- 现有蛋白质-配体共折叠方法受限于数据稀缺、架构低效和物理有效性不足,难以充分利用辅助信息。
- Pearl通过大规模合成数据训练、SO(3)-等变扩散模块和可控推理,提升蛋白质-配体共折叠的精度和效率。
- Pearl在多个基准测试中超越AlphaFold 3等模型,在真实药物靶标数据集上实现了显著的性能提升。
📝 摘要(中文)
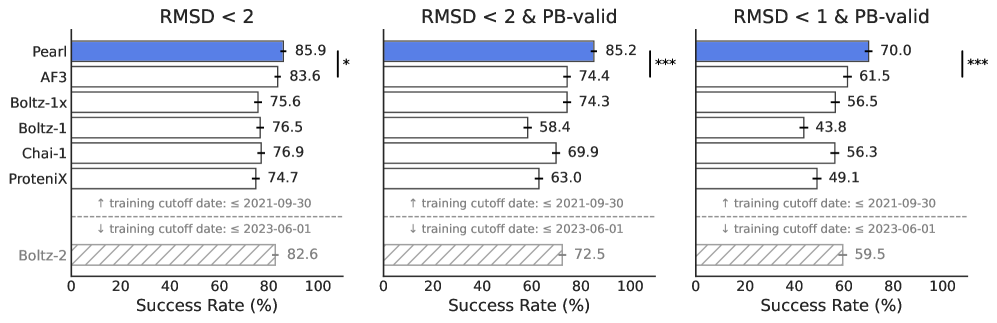
准确预测蛋白质-配体复合物的三维结构是计算药物发现中的一个基本挑战,它限制了治疗设计的速度和成功率。深度学习方法最近作为结构预测工具显示出强大的潜力,在不同的生物分子系统中实现了有希望的准确性。然而,它们的性能和效用受到稀缺的实验数据、低效的架构、物理上无效的姿势以及利用推理时可用的辅助信息的有限能力的限制。为了解决这些问题,我们引入了Pearl(将每个原子放置在正确的位置),这是一个用于大规模蛋白质-配体共折叠的基础模型。Pearl通过三个关键创新解决了这些挑战:(1)包括大规模合成数据的训练方案,以克服数据稀缺问题;(2)包含SO(3)-等变扩散模块的架构,以固有地尊重3D旋转对称性,从而提高泛化能力和样本效率;(3)可控推理,包括支持蛋白质和非聚合组分的通用多链模板系统以及双重无条件/条件模式。Pearl在蛋白质-配体共折叠方面建立了新的最先进的性能。在生成准确(RMSD < 2 Å)和物理上有效的姿势的关键指标上,Pearl超越了AlphaFold 3和其他开源基线,在公共Runs N' Poses和PoseBusters基准测试中分别实现了14.5%和14.2%的改进。在口袋条件共折叠方案中,Pearl在更严格的RMSD < 1 Å阈值下,在一组具有挑战性的真实药物靶标上实现了3.6倍的改进。最后,我们证明了模型性能与训练中使用的合成数据集大小直接相关。
🔬 方法详解
问题定义:蛋白质-配体共折叠是药物发现的关键步骤,但现有方法在预测复合物三维结构时面临数据稀缺、计算效率低和物理有效性不足等问题。这些问题限制了药物设计的速度和成功率。现有方法难以充分利用可用的辅助信息,例如口袋信息或模板结构。
核心思路:Pearl的核心思路是利用大规模合成数据来弥补实验数据的不足,并采用SO(3)-等变扩散模型来保证预测结构的物理合理性。通过可控推理,Pearl可以灵活地利用各种辅助信息,例如模板结构或口袋信息,从而提高预测精度。这种设计旨在克服现有方法的局限性,并实现更准确、更高效的蛋白质-配体共折叠。
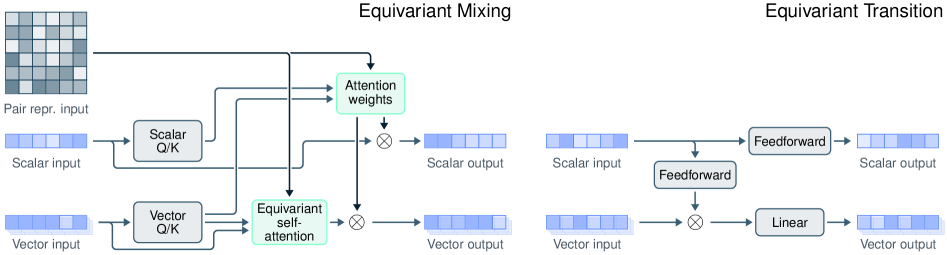
技术框架:Pearl的整体架构包含三个主要模块:大规模合成数据生成模块、SO(3)-等变扩散模块和可控推理模块。首先,生成大规模的合成蛋白质-配体复合物数据,用于模型的预训练。然后,利用SO(3)-等变扩散模块对蛋白质和配体的结构进行建模,保证预测结构的旋转不变性。最后,通过可控推理模块,可以灵活地利用各种辅助信息,例如模板结构或口袋信息,来指导结构的预测。
关键创新:Pearl的关键创新在于三个方面:(1) 大规模合成数据训练,克服了数据稀缺问题;(2) SO(3)-等变扩散模块,保证了预测结构的物理合理性;(3) 可控推理,可以灵活地利用各种辅助信息。与现有方法相比,Pearl能够更有效地利用数据,并生成更准确、更可靠的蛋白质-配体复合物结构。
关键设计:Pearl的关键设计包括:(1) 合成数据的生成策略,例如使用不同的蛋白质和配体库,并采用不同的对接方法生成复合物;(2) SO(3)-等变扩散模块的网络结构,例如使用球谐函数来表示原子坐标,并采用等变卷积操作;(3) 可控推理模块的实现方式,例如使用注意力机制来融合模板结构或口袋信息。
🖼️ 关键图片

📊 实验亮点
Pearl在Runs N' Poses和PoseBusters基准测试中,生成RMSD < 2 Å且物理有效的姿势的准确率分别提升了14.5%和14.2%,超越了AlphaFold 3和其他开源模型。在更严格的RMSD < 1 Å阈值下,Pearl在真实药物靶标数据集上实现了3.6倍的性能提升。模型性能与训练中使用的合成数据集大小直接相关。
🎯 应用场景
Pearl在药物发现领域具有广泛的应用前景,可用于加速先导化合物的发现、优化和筛选。通过准确预测蛋白质-配体复合物的结构,Pearl可以帮助研究人员更好地理解药物的作用机制,并设计出更有效、更安全的药物。此外,Pearl还可以应用于蛋白质工程、生物材料设计等领域。
📄 摘要(原文)
Accurately predicting the three-dimensional structures of protein-ligand complexes remains a fundamental challenge in computational drug discovery that limits the pace and success of therapeutic design. Deep learning methods have recently shown strong potential as structural prediction tools, achieving promising accuracy across diverse biomolecular systems. However, their performance and utility are constrained by scarce experimental data, inefficient architectures, physically invalid poses, and the limited ability to exploit auxiliary information available at inference. To address these issues, we introduce Pearl (Placing Every Atom in the Right Location), a foundation model for protein-ligand cofolding at scale. Pearl addresses these challenges with three key innovations: (1) training recipes that include large-scale synthetic data to overcome data scarcity; (2) architectures that incorporate an SO(3)-equivariant diffusion module to inherently respect 3D rotational symmetries, improving generalization and sample efficiency, and (3) controllable inference, including a generalized multi-chain templating system supporting both protein and non-polymeric components as well as dual unconditional/conditional modes. Pearl establishes a new state-of-the-art performance in protein-ligand cofolding. On the key metric of generating accurate (RMSD < 2 Å) and physically valid poses, Pearl surpasses AlphaFold 3 and other open source baselines on the public Runs N' Poses and PoseBusters benchmarks, delivering 14.5% and 14.2% improvements, respectively, over the next best model. In the pocket-conditional cofolding regime, Pearl delivers $3.6\times$ improvement on a proprietary set of challenging, real-world drug targets at the more rigorous RMSD < 1 Å threshold. Finally, we demonstrate that model performance correlates directly with synthetic dataset size used in training.