Foundation Models for Discovery and Exploration in Chemical Space
作者: Alexius Wadell, Anoushka Bhutani, Victor Azumah, Austin R. Ellis-Mohr, Celia Kelly, Hancheng Zhao, Anuj K. Nayak, Kareem Hegazy, Alexander Brace, Hongyi Lin, Murali Emani, Venkatram Vishwanath, Kevin Gering, Melisa Alkan, Tom Gibbs, Jack Wells, Lav R. Varshney, Bharath Ramsundar, Karthik Duraisamy, Michael W. Mahoney, Arvind Ramanathan, Venkatasubramanian Viswanathan
分类: physics.chem-ph, cond-mat.mtrl-sci, cs.LG
发布日期: 2025-10-20
备注: Main manuscript: 28 pages (including references), 7 tables and 5 figures. Supplementary information: 91 pages (including references), 12 tables and 82 figures
💡 一句话要点
MIST分子基石模型:助力化学空间探索与材料发现
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 分子基石模型 化学空间探索 材料发现 深度学习 分子性质预测
📋 核心要点
- 现有计算和实验方法在高效探索化学空间方面缺乏可扩展性,难以满足材料创新的需求。
- 论文提出MIST分子基石模型,通过新颖的token化方案和大规模数据训练,学习分子结构与性质之间的关系。
- 实验表明,MIST模型在多个化学性质预测任务中达到或超过了现有最佳性能,并能解决实际化学问题。
📝 摘要(中文)
本文提出了一系列分子基石模型MIST,其参数规模和训练数据量均比以往工作高出一个数量级。MIST采用一种新颖的token化方案,全面捕捉原子核、电子和几何信息,并从多样化的分子数据中学习。MIST模型经过微调后,能够预测超过400种结构-性质关系,并在生理学、电化学和量子化学等基准测试中达到或超过现有最佳性能。论文展示了这些模型解决化学空间中实际问题的能力,包括多目标电解质溶剂筛选、嗅觉感知映射、同位素半衰期预测、手性有机金属化合物的立体化学推理以及二元和多元混合物性质预测。通过可解释性方法探究MIST模型,揭示了训练数据中未明确存在的模式和趋势,表明该模型学习了可泛化的科学概念。论文还提出了超参数惩罚的贝叶斯神经缩放定律,并将模型开发计算成本降低了一个数量级。这些方法和发现代表了利用基石模型加速材料发现、设计和优化的重要一步,并为训练计算优化的科学基石模型提供了有价值的指导。
🔬 方法详解
问题定义:现有计算和实验方法在预测分子结构相关的原子、热力学和动力学性质时,缺乏足够的可扩展性,无法有效探索巨大的化学空间。这阻碍了新材料的发现和优化。
核心思路:论文的核心思路是利用大规模无标签数据训练科学基石模型,使其能够学习到分子结构与性质之间的潜在关系,从而实现对化学空间的高效探索。通过学习大量数据,模型能够泛化到未知的分子结构,并预测其性质。
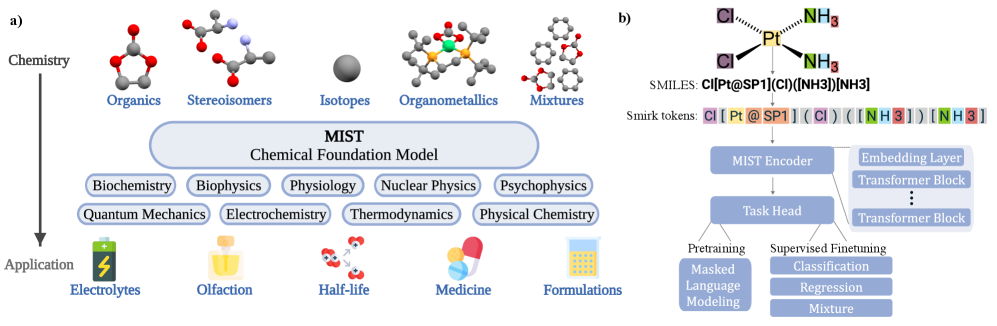
技术框架:MIST模型的整体框架包括数据预处理、token化、模型训练和微调等几个主要阶段。首先,对分子数据进行清洗和标准化。然后,使用一种新颖的token化方案,将分子结构信息转化为模型可以理解的token序列。接下来,使用大规模无标签数据训练模型,使其学习到分子结构与性质之间的关系。最后,使用少量有标签数据对模型进行微调,使其能够更好地预测特定性质。
关键创新:MIST模型的关键创新在于其token化方案和模型规模。该token化方案能够全面捕捉原子核、电子和几何信息,从而使模型能够更好地理解分子结构。此外,MIST模型具有比以往工作更大的参数规模和训练数据量,使其能够学习到更复杂的分子结构与性质之间的关系。
关键设计:MIST模型使用了Transformer架构,并针对分子数据的特点进行了优化。在token化方面,论文提出了一种新的方法,综合考虑了原子类型、键类型、原子坐标等信息。在损失函数方面,论文使用了多种损失函数,包括回归损失、分类损失和对比损失等,以提高模型的预测精度和泛化能力。此外,论文还提出了超参数惩罚的贝叶斯神经缩放定律,用于指导模型训练,降低计算成本。
🖼️ 关键图片
📊 实验亮点
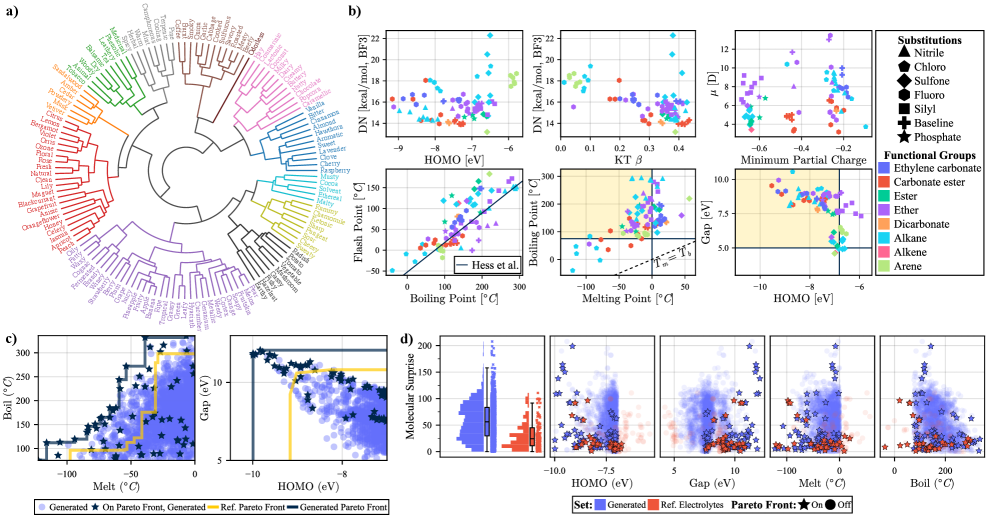
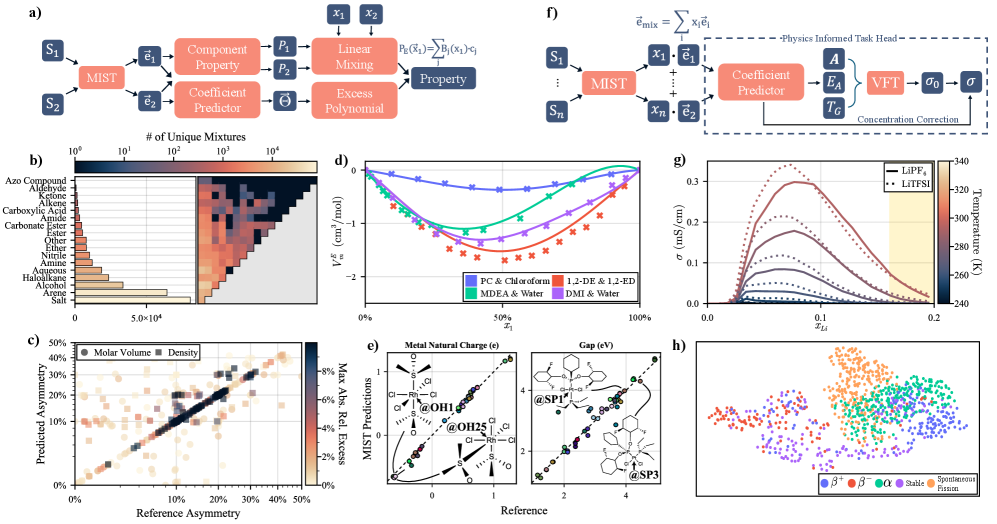
MIST模型在多个化学性质预测任务中取得了显著的成果。例如,在电解质溶剂筛选任务中,MIST模型能够有效地筛选出具有特定性质的溶剂。在嗅觉感知映射任务中,MIST模型能够准确地预测分子的气味。在同位素半衰期预测任务中,MIST模型能够准确地预测同位素的半衰期。此外,MIST模型还能够进行手性有机金属化合物的立体化学推理,并预测二元和多元混合物的性质。在多个基准测试中,MIST模型达到了或超过了现有最佳性能。
🎯 应用场景
MIST模型具有广泛的应用前景,可用于加速材料发现、设计和优化。例如,可以利用MIST模型筛选具有特定性质的电解质溶剂,预测新分子的气味,预测同位素的半衰期,以及进行手性有机金属化合物的立体化学推理。此外,MIST模型还可以用于预测二元和多元混合物的性质,从而为化学工程和材料科学提供有价值的指导。
📄 摘要(原文)
Accurate prediction of atomistic, thermodynamic, and kinetic properties from molecular structures underpins materials innovation. Existing computational and experimental approaches lack the scalability required to efficiently navigate chemical space. Scientific foundation models trained on large unlabeled datasets offer a path toward exploring chemical space across diverse application domains. Here we develop MIST, a family of molecular foundation models with up to an order of magnitude more parameters and data than prior works. Trained using a novel tokenization scheme that comprehensively captures nuclear, electronic, and geometric information, MIST learns from a diverse range of molecules. MIST models have been fine-tuned to predict more than 400 structure -- property relationships and match or exceed state-of-the-art performance across benchmarks spanning physiology, electrochemistry, and quantum chemistry. We demonstrate the ability of these models to solve real-world problems across chemical space, including multiobjective electrolyte solvent screening, olfactory perception mapping, isotope half-life prediction, stereochemical reasoning for chiral organometallic compounds, and binary and multi-component mixture property prediction. Probing MIST models using mechanistic interpretability methods reveals identifiable patterns and trends not explicitly present in the training data, suggesting that the models learn generalizable scientific concepts. We formulate hyperparameter-penalized Bayesian neural scaling laws and use them to reduce the computational cost of model development by an order of magnitude. The methods and findings presented here represent a significant step toward accelerating materials discovery, design, and optimization using foundation models and provide valuable guidance for training compute-optimal scientific foundation models.