Predictive Free Energy Simulations Through Hierarchical Distillation of Quantum Hamiltonians
作者: Chenghan Li, Garnet Kin-Lic Chan
分类: physics.chem-ph, cs.LG, physics.bio-ph
发布日期: 2025-09-13
💡 一句话要点
提出基于分层蒸馏量子哈密顿量的预测自由能模拟方法
🎯 匹配领域: 支柱二:RL算法与架构 (RL & Architecture)
关键词: 量子化学 机器学习 自由能模拟 分层蒸馏 量子哈密顿量
📋 核心要点
- 高精度量子力学计算在凝聚相反应自由能模拟中面临计算量巨大的挑战。
- 论文提出一种分层机器学习框架,通过蒸馏高精度量子计算结果,构建粗粒度的量子哈密顿量。
- 实验验证表明,该方法能够以化学精度重现弱酸解离常数和酶促反应速率的实验结果。
📝 摘要(中文)
对于高水平量子力学方法而言,获得凝聚相化学反应的自由能仍然在计算上是极其困难的。本文介绍了一种分层机器学习框架,通过将少量高保真量子计算的知识提炼成越来越粗粒度的、机器学习的量子哈密顿量,从而弥合了这一差距。通过保留显式的电子自由度,我们的方法进一步实现了量子和经典自由度的忠实嵌入,从而以无限阶捕获长程静电和量子对经典环境的响应。作为验证,我们完全从第一性原理计算了弱酸的质子解离常数和酶促反应的动力学速率,在化学精度或其不确定性范围内重现了实验测量结果。我们的工作展示了一条以最高的精度和收敛的统计数据进行反应自由能凝聚相模拟的途径。
🔬 方法详解
问题定义:论文旨在解决高精度量子力学方法在凝聚相化学反应自由能计算中计算成本过高的问题。现有的高精度量子计算方法,如耦合簇理论,虽然精度高,但计算量随体系尺寸呈指数级增长,难以应用于复杂体系的模拟。因此,如何降低计算成本,同时保持足够的精度,是该领域面临的关键挑战。
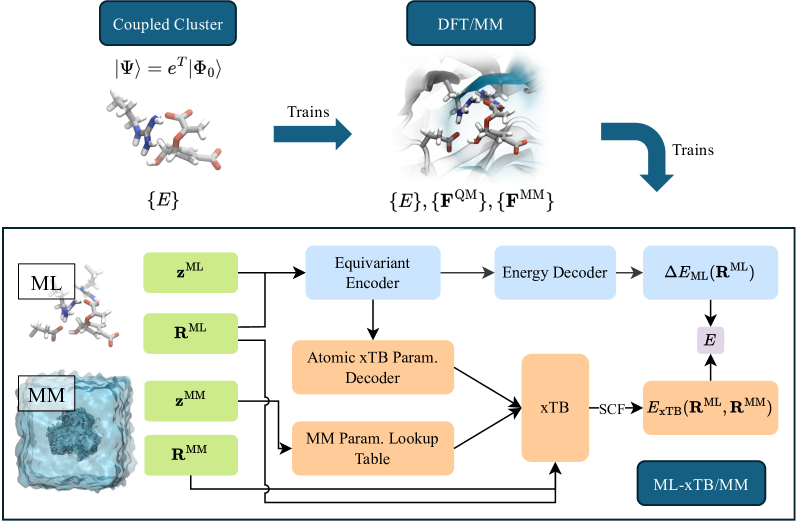
核心思路:论文的核心思路是通过分层蒸馏的方式,将少量高精度量子计算的结果迁移到计算成本更低的粗粒度模型中。具体而言,首先使用高精度量子力学方法计算少量具有代表性的构型,然后利用机器学习方法训练一个粗粒度的量子哈密顿量,该哈密顿量能够以较低的计算成本预测体系的能量和性质。通过多层蒸馏,逐步降低计算成本,最终实现对复杂体系的自由能模拟。
技术框架:该方法包含以下几个主要阶段:1) 高精度量子计算:使用高精度的量子力学方法(如耦合簇理论)计算少量具有代表性的构型的能量和性质。2) 特征提取:从高精度计算结果中提取关键的电子结构特征,例如原子电荷、键级等。3) 模型训练:使用机器学习方法(如神经网络)训练一个粗粒度的量子哈密顿量,该哈密顿量以提取的特征为输入,预测体系的能量和性质。4) 分层蒸馏:通过多层蒸馏,逐步降低计算成本,同时保持足够的精度。5) 自由能计算:使用训练好的粗粒度量子哈密顿量进行分子动力学模拟,并计算体系的自由能。
关键创新:该方法最重要的技术创新点在于分层蒸馏的思想,以及显式电子自由度的保留。分层蒸馏能够有效地降低计算成本,同时保持足够的精度。通过保留显式的电子自由度,该方法能够更好地描述体系的电子结构,从而提高模拟的准确性。与传统的基于力场的分子动力学模拟相比,该方法能够更好地描述化学反应过程中的电子结构变化。
关键设计:论文中关键的设计包括:1) 特征的选择:选择合适的电子结构特征对于训练一个准确的粗粒度量子哈密顿量至关重要。2) 机器学习模型的选择:选择合适的机器学习模型(如神经网络)对于提高预测精度至关重要。3) 损失函数的设计:设计合适的损失函数能够引导机器学习模型学习到正确的物理规律。4) 分层蒸馏的策略:选择合适的分层蒸馏策略能够有效地降低计算成本,同时保持足够的精度。
🖼️ 关键图片

📊 实验亮点
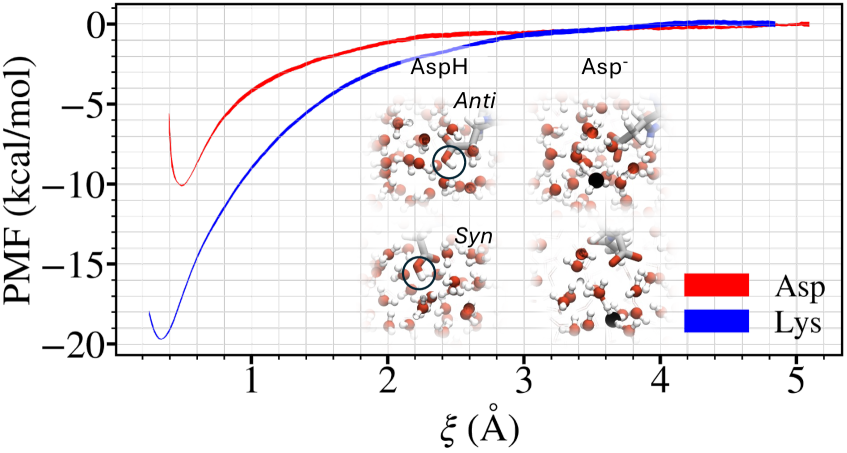
该论文通过第一性原理计算了弱酸的质子解离常数和酶促反应的动力学速率,结果与实验测量值在化学精度或其不确定性范围内吻合。这表明该方法能够以较高的精度预测凝聚相化学反应的性质,为复杂化学体系的模拟提供了一种新的途径。
🎯 应用场景
该研究成果可应用于凝聚相化学反应的模拟,例如催化反应、生物酶反应等。通过该方法,研究人员可以更准确地预测反应的自由能和动力学速率,从而加速新材料和新药物的开发。此外,该方法还可以应用于材料科学领域,例如预测材料的性质和结构。
📄 摘要(原文)
Obtaining the free energies of condensed phase chemical reactions remains computationally prohibitive for high-level quantum mechanical methods. We introduce a hierarchical machine learning framework that bridges this gap by distilling knowledge from a small number of high-fidelity quantum calculations into increasingly coarse-grained, machine-learned quantum Hamiltonians. By retaining explicit electronic degrees of freedom, our approach further enables a faithful embedding of quantum and classical degrees of freedom that captures long-range electrostatics and the quantum response to a classical environment to infinite order. As validation, we compute the proton dissociation constants of weak acids and the kinetic rate of an enzymatic reaction entirely from first principles, reproducing experimental measurements within chemical accuracy or their uncertainties. Our work demonstrates a path to condensed phase simulations of reaction free energies at the highest levels of accuracy with converged statistics.