DepMicroDiff: Diffusion-Based Dependency-Aware Multimodal Imputation for Microbiome Data
作者: Rabeya Tus Sadia, Qiang Cheng
分类: cs.LG, cs.CV
发布日期: 2025-07-31
💡 一句话要点
DepMicroDiff:结合依赖感知的扩散模型用于微生物组数据多模态补全
🎯 匹配领域: 支柱二:RL算法与架构 (RL & Architecture) 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 微生物组数据补全 扩散模型 依赖感知Transformer 多模态学习 癌症微生物组
📋 核心要点
- 现有微生物组数据补全方法难以捕捉微生物类群间的复杂依赖关系,且忽略了有用的上下文元数据。
- DepMicroDiff结合扩散模型与依赖感知Transformer,显式建模微生物间的成对依赖和自回归关系。
- 实验表明,DepMicroDiff在多种癌症类型微生物组数据补全任务上显著优于现有方法,提升了相关性和相似性。
📝 摘要(中文)
微生物组数据分析对于理解宿主健康和疾病至关重要,但其固有的稀疏性和噪声对准确的补全构成了重大挑战,阻碍了生物标志物发现等下游任务。现有的补全方法,包括最近基于扩散的模型,通常无法捕捉微生物类群之间复杂的相互依赖关系,并且忽略了可以为补全提供信息的上下文元数据。我们引入了DepMicroDiff,这是一个新颖的框架,它结合了基于扩散的生成模型和一个依赖感知Transformer(DAT),以显式地捕捉互惠的成对依赖关系和自回归关系。DepMicroDiff通过基于VAE的跨多个癌症数据集的预训练以及通过大型语言模型(LLM)编码的患者元数据进行调节,得到了进一步的增强。在TCGA微生物组数据集上的实验表明,DepMicroDiff显著优于最先进的基线,在多种癌症类型中实现了更高的Pearson相关性(高达0.712)、余弦相似性(高达0.812)以及更低的RMSE和MAE,证明了其微生物组补全的鲁棒性和泛化性。
🔬 方法详解
问题定义:微生物组数据分析面临数据稀疏性和噪声的挑战,导致下游任务(如生物标志物发现)的性能下降。现有的补全方法,包括基于扩散模型的,无法充分利用微生物类群之间的复杂依赖关系,并且忽略了患者元数据等上下文信息,限制了补全的准确性。
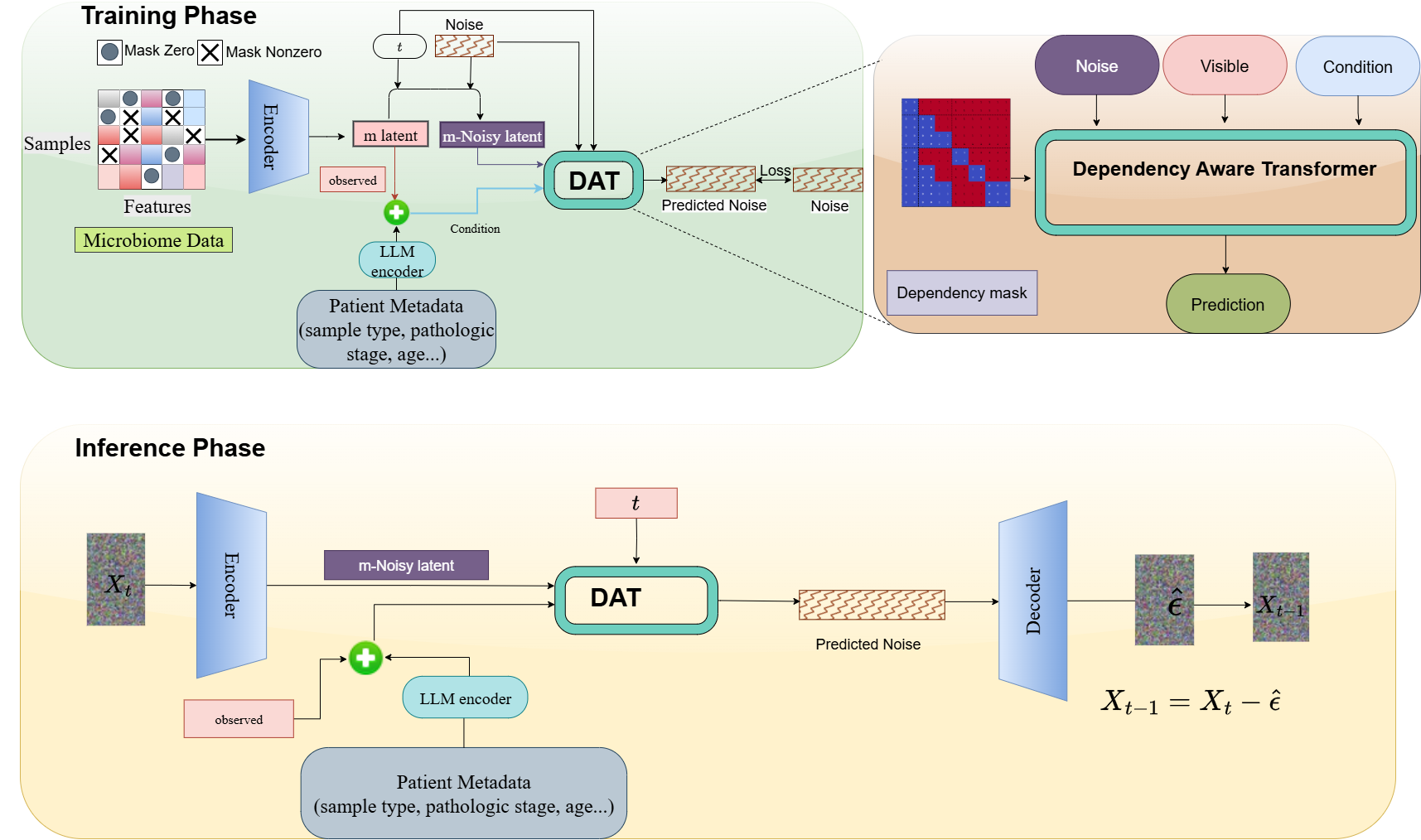
核心思路:DepMicroDiff的核心思路是将扩散模型与依赖感知Transformer(DAT)相结合,显式地建模微生物类群之间的成对依赖关系和自回归关系。通过DAT学习微生物之间的依赖关系,并将其融入到扩散模型的生成过程中,从而提高补全的准确性和鲁棒性。同时,利用VAE预训练和LLM编码的患者元数据作为条件,进一步提升补全效果。
技术框架:DepMicroDiff的整体框架包括以下几个主要模块:1) VAE预训练模块:使用VAE在多个癌症数据集上进行预训练,学习微生物组数据的潜在表示。2) 依赖感知Transformer(DAT):用于学习微生物类群之间的依赖关系,输出依赖关系矩阵。3) 扩散模型:基于扩散过程生成完整的微生物组数据,DAT提供的依赖关系矩阵作为扩散过程的调节信息。4) LLM编码器:将患者元数据编码为向量表示,作为扩散模型的条件输入。
关键创新:DepMicroDiff的关键创新在于:1) 提出了依赖感知Transformer(DAT),能够有效地学习微生物类群之间的复杂依赖关系。2) 将DAT与扩散模型相结合,利用学习到的依赖关系指导扩散过程,从而提高补全的准确性。3) 结合VAE预训练和LLM编码的患者元数据,进一步提升了补全效果。与现有方法相比,DepMicroDiff能够更有效地利用微生物组数据中的依赖关系和上下文信息。
关键设计:DAT采用Transformer结构,输入为微生物组数据,输出为依赖关系矩阵。扩散模型采用标准的扩散模型结构,损失函数包括扩散损失和重构损失。VAE的结构为标准的编码器-解码器结构,损失函数包括重构损失和KL散度。LLM采用预训练的BERT模型,将患者元数据编码为向量表示。
🖼️ 关键图片

📊 实验亮点
DepMicroDiff在TCGA微生物组数据集上进行了广泛的实验,结果表明,DepMicroDiff显著优于现有的补全方法。具体而言,DepMicroDiff在多种癌症类型中实现了更高的Pearson相关性(高达0.712)、余弦相似性(高达0.812)以及更低的RMSE和MAE。这些结果表明,DepMicroDiff具有很强的鲁棒性和泛化性,能够有效地应用于不同的微生物组数据集。
🎯 应用场景
DepMicroDiff可应用于多种微生物组数据分析场景,例如:疾病诊断、生物标志物发现、个性化医疗等。通过准确补全缺失的微生物组数据,可以提高下游分析的准确性和可靠性,从而更好地理解微生物与宿主健康之间的关系,为疾病的预防和治疗提供新的思路。
📄 摘要(原文)
Microbiome data analysis is essential for understanding host health and disease, yet its inherent sparsity and noise pose major challenges for accurate imputation, hindering downstream tasks such as biomarker discovery. Existing imputation methods, including recent diffusion-based models, often fail to capture the complex interdependencies between microbial taxa and overlook contextual metadata that can inform imputation. We introduce DepMicroDiff, a novel framework that combines diffusion-based generative modeling with a Dependency-Aware Transformer (DAT) to explicitly capture both mutual pairwise dependencies and autoregressive relationships. DepMicroDiff is further enhanced by VAE-based pretraining across diverse cancer datasets and conditioning on patient metadata encoded via a large language model (LLM). Experiments on TCGA microbiome datasets show that DepMicroDiff substantially outperforms state-of-the-art baselines, achieving higher Pearson correlation (up to 0.712), cosine similarity (up to 0.812), and lower RMSE and MAE across multiple cancer types, demonstrating its robustness and generalizability for microbiome imputation.