Distilling and exploiting quantitative insights from Large Language Models for enhanced Bayesian optimization of chemical reactions
作者: Roshan Patel, Saeed Moayedpour, Louis De Lescure, Lorenzo Kogler-Anele, Alan Cherney, Sven Jager, Yasser Jangjou
分类: cs.LG, cs.AI
发布日期: 2025-04-11
💡 一句话要点
利用大语言模型知识蒸馏,增强化学反应贝叶斯优化
🎯 匹配领域: 支柱二:RL算法与架构 (RL & Architecture) 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 贝叶斯优化 大语言模型 化学反应优化 迁移学习 知识蒸馏 偏好学习 提示工程
📋 核心要点
- 化学反应优化面临数据稀缺挑战,传统贝叶斯优化方法难以充分利用先验知识。
- 利用大语言模型蕴含的化学知识,通过提示工程和偏好学习构建效用函数,指导贝叶斯优化。
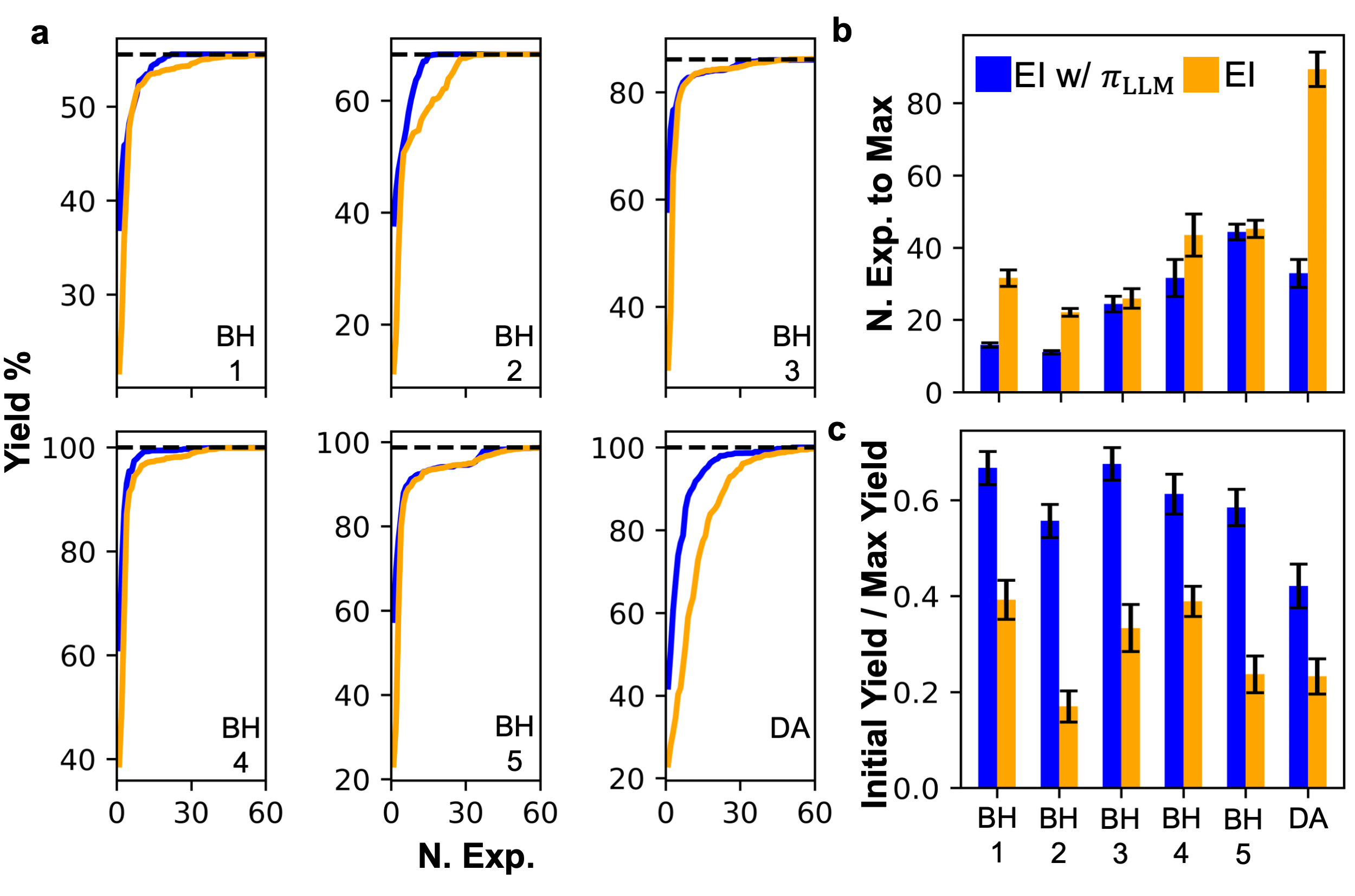
- 实验表明,该方法能有效聚焦优化过程,在多个数据集上提升了贝叶斯优化的初始产率和整体性能。
📝 摘要(中文)
机器学习和贝叶斯优化(BO)算法能显著加速化学反应优化。迁移学习通过利用已有的化学信息或优化任务之外的数据(即源数据)来增强BO算法在低数据情况下的有效性。大语言模型(LLM)已经证明,基础训练数据中存在的化学信息使其能够处理化学数据。此外,它们可以被增强,并有助于合成与优化任务相关的潜在的多种源化学数据。本文研究了如何从LLM中提取化学信息,并将其用于迁移学习,以加速反应条件的BO,从而最大化产率。具体来说,我们表明,类似于调查的提示方案和偏好学习可以用来推断一个效用函数,该函数对LLM中嵌入的化学参数空间上的先验化学信息进行建模;我们发现,尽管在零样本设置下运行,但效用函数与参数空间上的真实实验测量值(产率)显示出适度的相关性。此外,我们表明,效用函数可以被用来将BO工作集中在有希望的参数空间区域,从而提高初始BO查询的产率,并增强在研究的6个数据集中的4个数据集上的优化。总的来说,我们认为这项工作是弥合LLM中嵌入的化学知识与有原则的BO方法加速反应优化能力之间差距的一步。
🔬 方法详解
问题定义:化学反应优化旨在寻找最佳反应条件以最大化产率。传统贝叶斯优化方法在数据量较少时表现不佳,难以有效利用已有的化学知识。现有方法缺乏有效利用大语言模型中蕴含的丰富化学信息的途径。
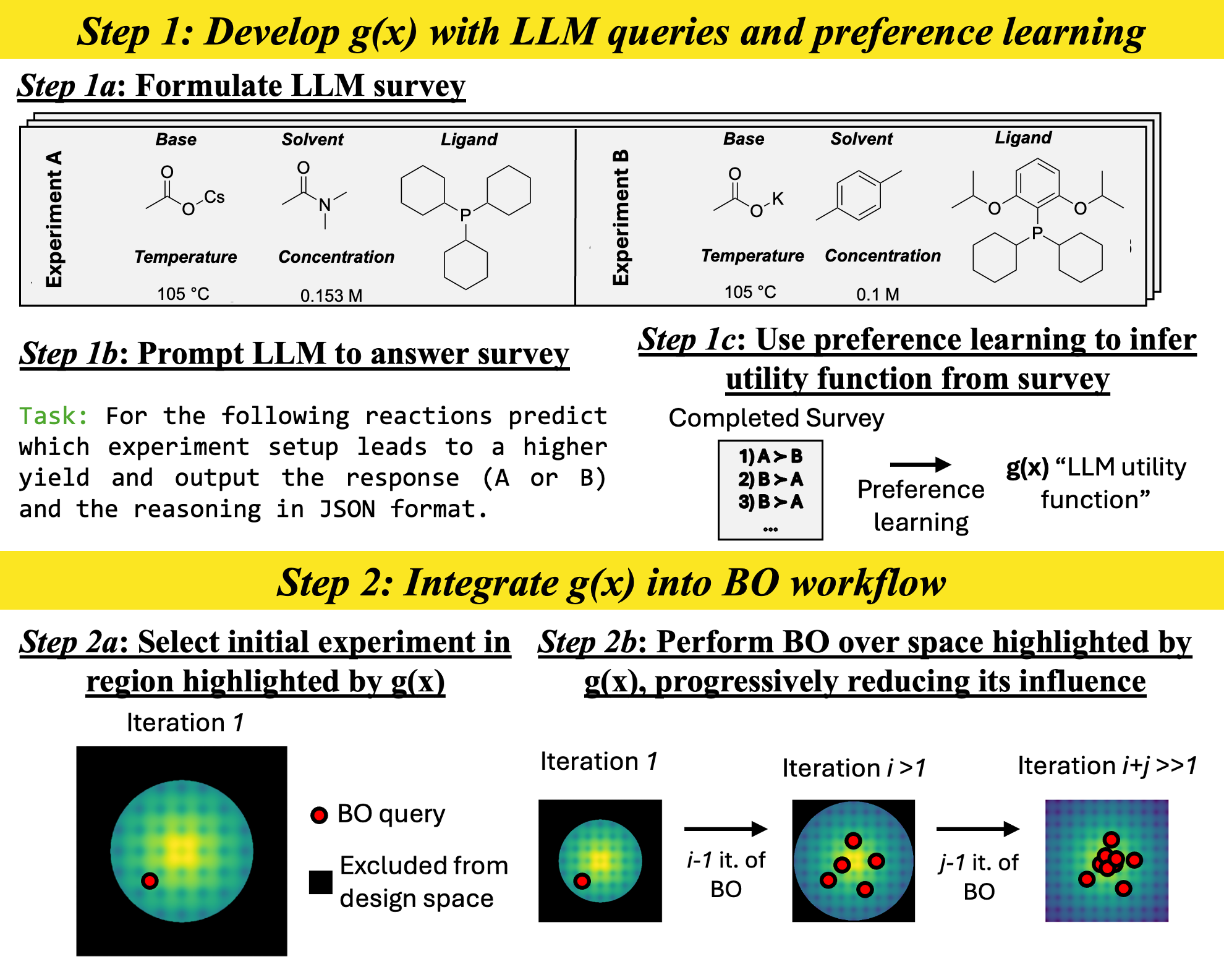
核心思路:本研究的核心思路是利用大语言模型(LLM)中蕴含的化学知识,通过知识蒸馏的方式,提取出对反应优化有用的先验信息,并将其融入到贝叶斯优化过程中。具体来说,通过设计合适的提示(prompt),引导LLM对不同反应条件进行排序,从而学习到一个效用函数,该函数能够反映LLM对反应产率的预测。
技术框架:整体框架包含以下几个主要阶段:1) LLM知识提取:设计调查式提示,向LLM询问不同反应参数组合的偏好排序。2) 效用函数构建:利用偏好学习算法,将LLM的排序结果转化为一个效用函数,该函数能够预测不同反应参数组合的产率。3) 贝叶斯优化:将效用函数作为先验知识融入到贝叶斯优化过程中,引导优化算法在更有希望的区域进行探索。
关键创新:本研究的关键创新在于:1) 利用LLM进行化学知识蒸馏:首次探索了利用LLM中蕴含的化学知识来指导化学反应优化。2) 调查式提示和偏好学习:提出了一种有效的提示工程方法,能够从LLM中提取出有用的先验信息。3) 将LLM知识融入贝叶斯优化:将提取的知识以效用函数的形式融入到贝叶斯优化过程中,提高了优化效率。
关键设计:1) 提示设计:设计类似于调查问卷的提示,例如“在以下反应条件下,哪个产率更高?”,并提供不同的反应参数组合供LLM选择。2) 偏好学习算法:使用基于排序的损失函数,训练一个模型来预测LLM对不同反应参数组合的偏好。3) 贝叶斯优化算法:使用高斯过程作为代理模型,并结合效用函数来选择下一个要评估的反应参数组合。
🖼️ 关键图片

📊 实验亮点
实验结果表明,该方法在6个数据集中的4个数据集上提高了贝叶斯优化的性能。即使在零样本设置下,从LLM提取的效用函数也与真实实验测量值(产率)表现出适度的相关性。该方法能够提高初始BO查询的产率,并增强整体优化效果。
🎯 应用场景
该研究成果可应用于加速新化学反应的发现和优化,降低实验成本,缩短研发周期。潜在应用领域包括药物研发、材料科学、精细化工等。通过结合大语言模型的知识和贝叶斯优化算法,有望实现更高效、智能的化学反应优化。
📄 摘要(原文)
Machine learning and Bayesian optimization (BO) algorithms can significantly accelerate the optimization of chemical reactions. Transfer learning can bolster the effectiveness of BO algorithms in low-data regimes by leveraging pre-existing chemical information or data outside the direct optimization task (i.e., source data). Large language models (LLMs) have demonstrated that chemical information present in foundation training data can give them utility for processing chemical data. Furthermore, they can be augmented with and help synthesize potentially multiple modalities of source chemical data germane to the optimization task. In this work, we examine how chemical information from LLMs can be elicited and used for transfer learning to accelerate the BO of reaction conditions to maximize yield. Specifically, we show that a survey-like prompting scheme and preference learning can be used to infer a utility function which models prior chemical information embedded in LLMs over a chemical parameter space; we find that the utility function shows modest correlation to true experimental measurements (yield) over the parameter space despite operating in a zero-shot setting. Furthermore, we show that the utility function can be leveraged to focus BO efforts in promising regions of the parameter space, improving the yield of the initial BO query and enhancing optimization in 4 of the 6 datasets studied. Overall, we view this work as a step towards bridging the gap between the chemistry knowledge embedded in LLMs and the capabilities of principled BO methods to accelerate reaction optimization.