Chemist-aligned retrosynthesis by ensembling diverse inductive bias models
作者: Krzysztof Maziarz, Guoqing Liu, Hubert Misztela, Austin Tripp, Junren Li, Aleksei Kornev, Piotr Gaiński, Holger Hoefling, Mike Fortunato, Rishi Gupta, Marwin Segler
分类: cs.LG, cs.AI, q-bio.QM
发布日期: 2024-12-06 (更新: 2025-08-12)
💡 一句话要点
RetroChimera:通过集成多样归纳偏置模型实现化学家对齐的逆合成
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 逆合成 化学合成 人工智能 集成学习 Transformer 图神经网络 药物发现 零样本迁移
📋 核心要点
- 现有AI逆合成模型在罕见反应和错误预测上表现不佳,阻碍多步搜索并与化学家期望不符。
- RetroChimera通过融合具有互补归纳偏置的两个新组件,并使用学习型集成策略整合预测来解决上述问题。
- 实验表明,RetroChimera在数据规模和分割策略上优于现有模型,并展现了良好的泛化能力和化学家对齐性。
📝 摘要(中文)
化学合成仍然是功能性小分子发现和制造的关键瓶颈。基于人工智能的合成规划模型有望找到有效的合成路线,近年来取得了进展。然而,它们在合成策略中不太常见但至关重要的反应以及虚构的、不正确的预测方面仍然存在困难。这阻碍了依赖模型的的多步搜索算法,并导致与化学家期望的不一致。本文提出了RetroChimera:一种前沿的逆合成模型,它建立在两个新开发的具有互补归纳偏置的组件之上,我们使用一种新的框架,通过基于学习的集成策略来整合来自多个来源的预测,从而将它们融合在一起。通过跨越多个数量级的数据规模和分割策略的实验,我们表明RetroChimera大大优于所有主要模型,证明了在训练数据之外的鲁棒性,并且首次展示了从每个反应类别中极少量示例中学习的能力。此外,工业有机化学家更喜欢RetroChimera的预测,而不是它所训练的反应,这表明了高度的一致性。最后,我们展示了零样本迁移到一家大型制药公司的内部数据集,显示了在分布偏移下的强大泛化能力。凭借我们的集成框架所解锁的新维度,我们预计将进一步加速开发更准确的模型。
🔬 方法详解
问题定义:论文旨在解决AI逆合成模型在处理罕见反应、产生错误预测以及与化学家期望不一致的问题。现有方法在这些方面存在不足,限制了其在实际化学合成规划中的应用。
核心思路:论文的核心思路是利用集成学习,将具有不同归纳偏置的多个模型进行融合,从而综合利用不同模型的优势,提高逆合成预测的准确性和可靠性。通过学习不同模型的预测结果,并根据其在不同情况下的表现进行加权,可以有效降低错误预测的概率,并更好地模拟化学家的合成策略。
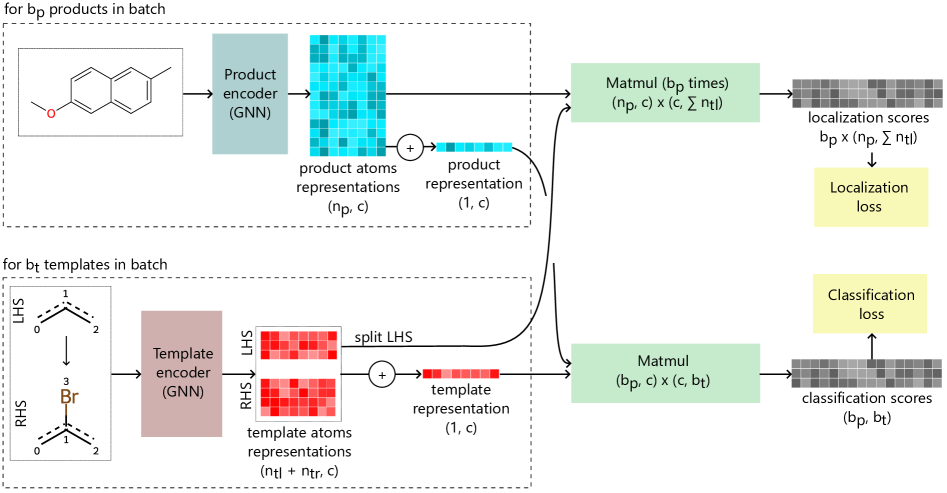
技术框架:RetroChimera模型包含两个主要组件:一个是基于Transformer的序列到序列模型,另一个是基于图神经网络的模型。这两个模型具有不同的归纳偏置,分别擅长捕捉反应物和产物的序列信息以及分子结构的拓扑信息。此外,该模型还包含一个学习型集成模块,用于学习如何将两个模型的预测结果进行融合。整个流程包括:输入目标分子,两个模型分别进行逆合成预测,集成模块根据模型的预测结果生成最终的逆合成反应。
关键创新:该论文的关键创新在于提出了一种新的学习型集成框架,用于融合具有不同归纳偏置的逆合成模型。与传统的模型集成方法相比,该框架能够根据不同反应的特点,动态地调整不同模型的权重,从而实现更准确的预测。此外,该模型还首次展示了从每个反应类别中极少量示例中学习的能力。
关键设计:集成模块使用一个神经网络来学习不同模型的权重。该网络的输入是两个模型的预测结果,输出是每个模型的权重。损失函数采用交叉熵损失函数,用于衡量预测结果与真实反应之间的差异。模型的训练数据包括大量的逆合成反应,以及化学家的合成策略。
🖼️ 关键图片

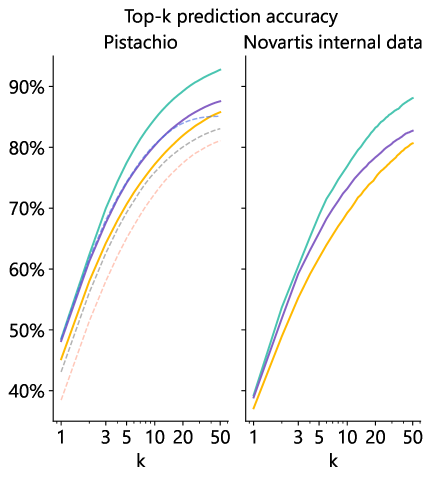
📊 实验亮点
RetroChimera在多个数据集上取得了显著的性能提升,大幅超越了现有的逆合成模型。在内部数据集上的零样本迁移实验表明,该模型具有很强的泛化能力。此外,工业有机化学家更倾向于RetroChimera的预测结果,表明该模型与化学家的合成策略高度一致。实验结果表明,RetroChimera能够有效地解决现有模型在罕见反应和错误预测方面的问题。
🎯 应用场景
该研究成果可应用于药物发现、材料科学等领域,加速新分子和材料的合成路线设计。通过提供更准确、可靠的逆合成预测,RetroChimera能够帮助化学家更高效地进行合成规划,降低实验成本,并发现新的合成路径。该模型还可用于自动化合成平台的开发,实现合成过程的智能化。
📄 摘要(原文)
Chemical synthesis remains a critical bottleneck in the discovery and manufacture of functional small molecules. AI-based synthesis planning models could be a potential remedy to find effective syntheses, and have made progress in recent years. However, they still struggle with less frequent, yet critical reactions for synthetic strategy, as well as hallucinated, incorrect predictions. This hampers multi-step search algorithms that rely on models, and leads to misalignment with chemists' expectations. Here we propose RetroChimera: a frontier retrosynthesis model, built upon two newly developed components with complementary inductive biases, which we fuse together using a new framework for integrating predictions from multiple sources via a learning-based ensembling strategy. Through experiments across several orders of magnitude in data scale and splitting strategy, we show RetroChimera outperforms all major models by a large margin, demonstrating robustness outside the training data, as well as for the first time the ability to learn from even a very small number of examples per reaction class. Moreover, industrial organic chemists prefer predictions from RetroChimera over the reactions it was trained on in terms of quality, revealing high levels of alignment. Finally, we demonstrate zero-shot transfer to an internal dataset from a major pharmaceutical company, showing robust generalization under distribution shift. With the new dimension that our ensembling framework unlocks, we anticipate further acceleration in the development of even more accurate models.