Large Language Model-Guided Prediction Toward Quantum Materials Synthesis
作者: Ryotaro Okabe, Zack West, Abhijatmedhi Chotrattanapituk, Mouyang Cheng, Denisse Córdova Carrizales, Weiwei Xie, Robert J. Cava, Mingda Li
分类: cond-mat.mtrl-sci, cs.LG
发布日期: 2024-10-28
备注: 66 pages total, 6 main figures + 3 supplementary figures
💡 一句话要点
利用大语言模型预测量子材料合成路径,加速新材料发现
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 大语言模型 量子材料 材料合成 化学方程式预测 广义谷本相似性
📋 核心要点
- 无机材料合成依赖大量实验试错,设计高效合成流程面临挑战。
- 利用大语言模型预测反应物、产物和完整化学方程式,指导材料合成。
- 通过广义谷本相似性微调,模型准确率提升至90%左右,且对量子材料有效。
📝 摘要(中文)
无机晶体材料的合成对现代技术至关重要,尤其是在量子材料的开发中。然而,由于精确的实验条件和大量的试错,设计高效的合成流程仍然是一个巨大的挑战。本文提出了一个使用大语言模型(LLM)来预测无机材料(包括量子材料)合成路径的框架。该框架包含三个模型:LHS2RHS(从反应物预测产物)、RHS2LHS(从产物预测反应物)和TGT2CEQ(为目标化合物生成完整的化学方程式)。该模型在一个文本挖掘的合成数据库上进行了微调,使用预训练模型的准确率低于40%,使用传统微调方法提高到低于80%,而使用本文提出的广义谷本相似性方法进一步提高到90%左右,同时保持了对额外合成步骤的鲁棒性。该模型还展示了在不同量子权重材料上的可比性能,表明LLM为预测量子材料发现的平衡化学方程式提供了一个强大的工具。
🔬 方法详解
问题定义:目前无机材料的合成,特别是量子材料的合成,高度依赖于实验经验和大量的试错,缺乏有效的理论指导。现有的方法难以准确预测合成反应的路径和条件,导致研发周期长、成本高。因此,如何利用已有的合成数据,构建一个能够准确预测合成路径的模型,是亟待解决的问题。
核心思路:本文的核心思路是利用大语言模型(LLM)强大的文本理解和生成能力,将化学合成过程视为一种文本翻译任务。通过训练LLM学习反应物和产物之间的关系,以及化学方程式的平衡规则,从而实现对合成路径的预测。这种方法避免了传统方法中复杂的物理化学建模,而是直接从数据中学习规律。
技术框架:该框架包含三个主要模型:LHS2RHS、RHS2LHS和TGT2CEQ。LHS2RHS模型用于从反应物预测产物,RHS2LHS模型用于从产物预测反应物,TGT2CEQ模型用于生成目标化合物的完整化学方程式。整个流程首先使用文本挖掘技术构建一个合成数据库,然后使用该数据库对LLM进行微调。在预测合成路径时,可以根据需要选择不同的模型组合,例如,可以先使用RHS2LHS模型预测反应物,然后使用LHS2RHS模型验证预测结果。
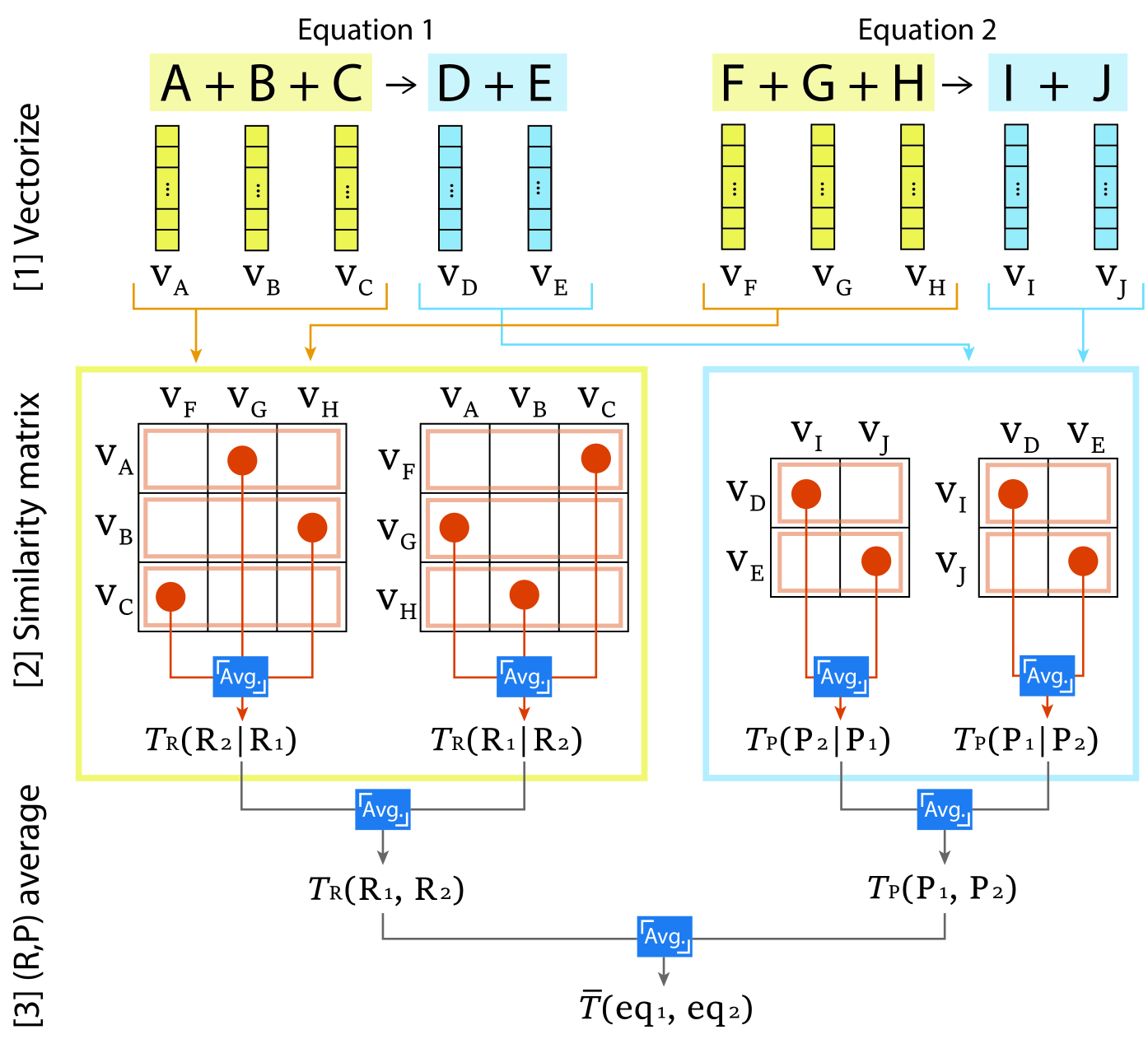
关键创新:本文最重要的技术创新点在于提出了广义谷本相似性(Generalized Tanimoto Similarity)用于微调LLM。传统的微调方法通常使用交叉熵损失函数,但这种方法忽略了化学方程式之间的相似性。广义谷本相似性能够衡量不同化学方程式之间的相似程度,从而使模型能够更好地学习合成反应的规律。与现有方法相比,该方法能够显著提高模型的预测准确率。
关键设计:在模型训练过程中,作者使用了文本挖掘技术构建了一个包含大量合成反应的数据库。为了提高模型的泛化能力,作者还对数据进行了增强处理。在损失函数方面,除了传统的交叉熵损失函数外,还引入了广义谷本相似性损失函数。具体而言,广义谷本相似性被用于衡量预测的化学方程式与真实化学方程式之间的相似度,并将其作为损失函数的一部分,从而引导模型学习更加合理的合成路径。
🖼️ 关键图片


📊 实验亮点
实验结果表明,使用预训练LLM的准确率低于40%,使用传统微调方法提高到低于80%,而使用本文提出的广义谷本相似性方法进一步提高到90%左右。该模型在不同量子权重的材料上表现出可比的性能,验证了其在量子材料合成预测方面的有效性。
🎯 应用场景
该研究成果可应用于新材料的快速发现和合成路径优化,尤其是在量子材料领域。通过LLM预测,研究人员可以减少试错次数,加速材料研发进程,降低研发成本。此外,该方法还可以用于指导自动化合成设备的运行,实现高通量材料合成。
📄 摘要(原文)
The synthesis of inorganic crystalline materials is essential for modern technology, especially in quantum materials development. However, designing efficient synthesis workflows remains a significant challenge due to the precise experimental conditions and extensive trial and error. Here, we present a framework using large language models (LLMs) to predict synthesis pathways for inorganic materials, including quantum materials. Our framework contains three models: LHS2RHS, predicting products from reactants; RHS2LHS, predicting reactants from products; and TGT2CEQ, generating full chemical equations for target compounds. Fine-tuned on a text-mined synthesis database, our model raises accuracy from under 40% with pretrained models, to under 80% using conventional fine-tuning, and further to around 90% with our proposed generalized Tanimoto similarity, while maintaining robust to additional synthesis steps. Our model further demonstrates comparable performance across materials with varying degrees of quantumness quantified using quantum weight, indicating that LLMs offer a powerful tool to predict balanced chemical equations for quantum materials discovery.