AI-Guided Molecular Simulations in VR: Exploring Strategies for Imitation Learning in Hyperdimensional Molecular Systems
作者: Mohamed Dhouioui, Jonathan Barnoud, Rhoslyn Roebuck Williams, Harry J. Stroud, Phil Bates, David R. Glowacki
分类: cs.LG, cs.AI, cs.HC, q-bio.BM
发布日期: 2024-09-11 (更新: 2025-11-03)
备注: (First presented at the First Workshop on "eXtended Reality \& Intelligent Agents" (XRIA24) @ ECAI24, Santiago De Compostela (Spain), 20 October 2024)
期刊: SN COMPUT. SCI. 6, 922 (2025)
DOI: 10.1007/s42979-025-04465-5
💡 一句话要点
利用VR中的AI引导分子模拟:探索超高维分子系统中模仿学习的策略
🎯 匹配领域: 支柱一:机器人控制 (Robot Control) 支柱二:RL算法与架构 (RL & Architecture)
关键词: 分子动力学模拟 虚拟现实 人机协同 模仿学习 AI智能体 分子操作 卷积神经网络
📋 核心要点
- 分子动力学模拟计算成本高昂,交互式分子动力学在虚拟现实(iMD-VR)中提供了一种人机协同策略,但缺乏自动化方法。
- 利用iMD-VR生成的数据集,通过模仿学习训练AI智能体,使智能体能够模仿专家操作,从而自动化分子操作任务。
- 通过初步研究,使用iMD-VR数据训练CNN网络,成功完成了一个简单的分子操作任务,验证了该方法的可行性。
📝 摘要(中文)
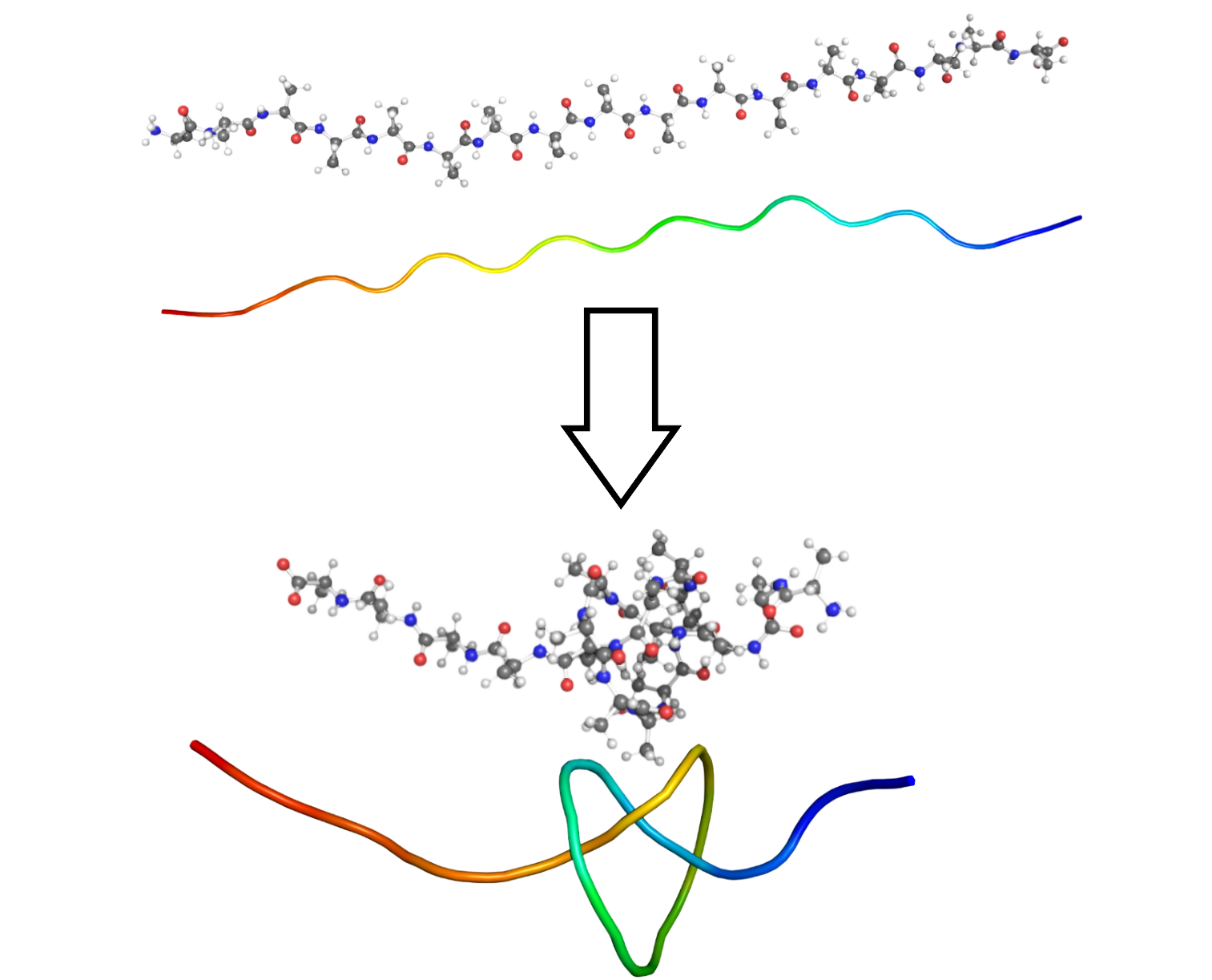
分子动力学(MD)模拟是研究人员理解和设计分子结构与功能的关键计算工具,广泛应用于药物发现、蛋白质工程和材料设计等领域。然而,由于分子系统的高维度特性,MD模拟计算成本高昂。虚拟现实中的交互式分子动力学(iMD-VR)作为一种“人机协同”策略,能够高效地导航超高维分子系统。iMD-VR提供沉浸式3D环境,实现对高性能计算架构上运行的实时分子模拟的可视化和操作,使研究人员能够引导分子构象动力学,从而高效探索复杂的高维分子系统。此外,iMD-VR模拟生成的数据集能够捕捉人类专家关于分子结构和功能的空间洞察力。本文探讨了利用研究人员生成的iMD-VR数据集,通过模仿学习(IL)训练AI智能体。IL使智能体能够模仿专家演示的复杂行为,避免了显式编程或复杂奖励函数设计。本文回顾了机器人和多智能体系统领域中与iMD-VR相似的IL应用,并讨论了如何使用iMD-VR记录来训练IL模型以与MD模拟交互。通过一个初步研究,展示了这些想法的应用,其中iMD-VR数据被用于训练CNN网络来完成一个简单的分子操作任务:将小分子穿过纳米管孔。最后,概述了未来研究方向以及使用AI智能体增强人类在导航巨大分子构象空间中的专业知识的潜在挑战。
🔬 方法详解
问题定义:论文旨在解决分子动力学模拟中,人工引导分子操作效率低下的问题。现有方法依赖于人工经验,缺乏自动化手段,难以应对复杂分子系统的探索。
核心思路:利用人类专家在iMD-VR环境中进行分子操作的数据,通过模仿学习训练AI智能体,使智能体能够学习并模仿专家的操作策略,从而实现分子操作的自动化。这种方法避免了手动设计复杂的奖励函数,直接从专家数据中学习。
技术框架:整体框架包含三个主要部分:1) iMD-VR环境:用于生成人类专家操作分子动力学模拟的数据集。2) 模仿学习模型:使用iMD-VR数据训练AI智能体,使其能够模仿专家的分子操作行为。3) 分子动力学模拟环境:AI智能体在此环境中与分子系统进行交互,执行分子操作任务。
关键创新:该研究的关键创新在于将模仿学习应用于iMD-VR生成的分子动力学模拟数据,从而实现分子操作的自动化。与传统的基于规则或优化的分子操作方法相比,该方法能够从人类专家的经验中学习,更有效地探索复杂的分子系统。
关键设计:在初步研究中,使用卷积神经网络(CNN)作为模仿学习模型,输入是iMD-VR环境中的分子状态信息,输出是智能体的操作指令。损失函数采用行为克隆(Behavior Cloning)方法,即最小化智能体操作与专家操作之间的差异。具体的网络结构和参数设置未在摘要中详细说明,属于未知信息。
🖼️ 关键图片

📊 实验亮点
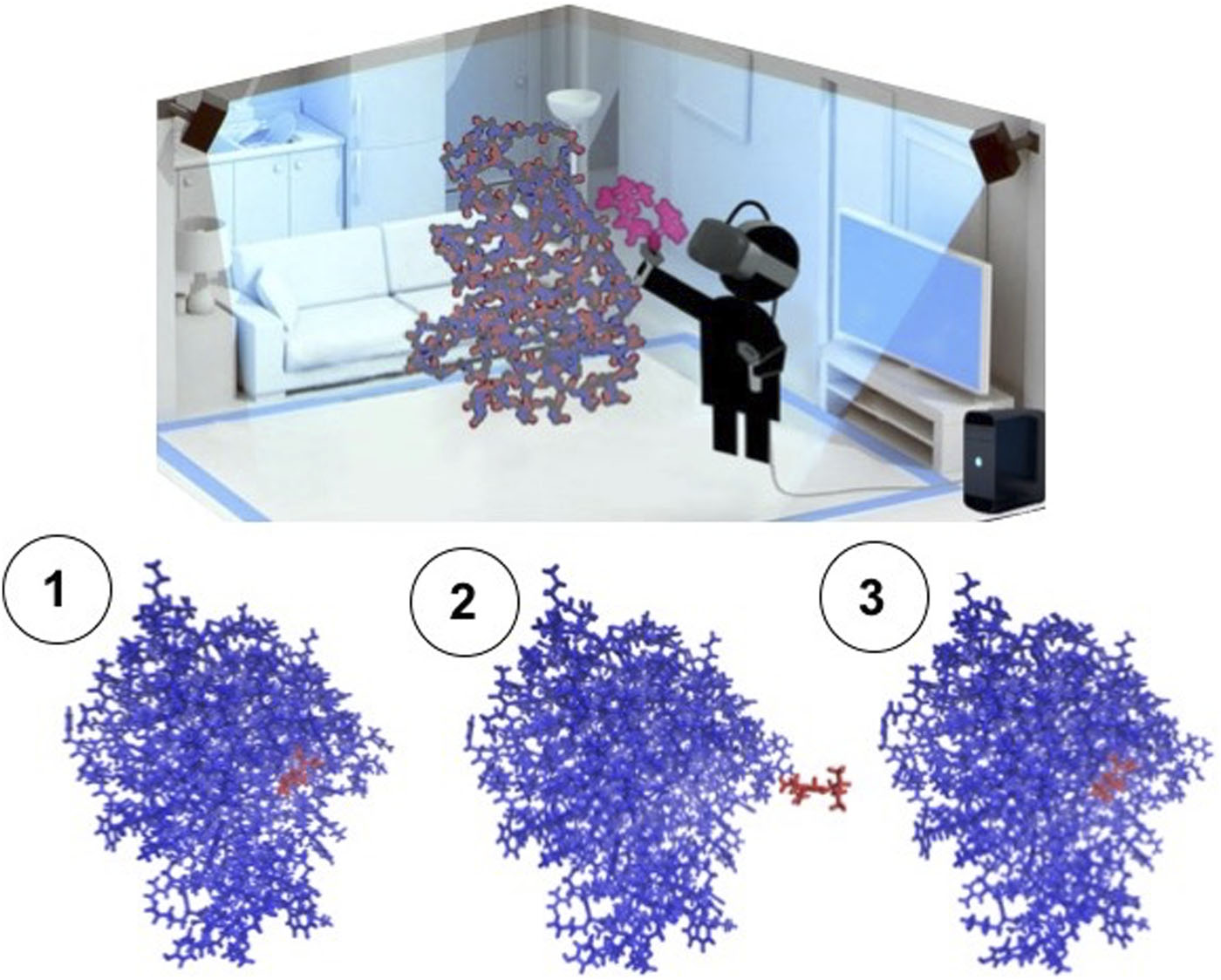
该研究通过初步实验,验证了使用iMD-VR数据训练CNN网络来完成分子操作任务的可行性。具体而言,成功训练了一个AI智能体,使其能够将小分子穿过纳米管孔。虽然摘要中没有提供具体的性能数据和对比基线,但该实验结果表明,模仿学习在分子动力学模拟领域具有巨大的应用潜力。
🎯 应用场景
该研究成果可应用于药物发现、蛋白质工程和材料设计等领域。通过AI智能体的辅助,研究人员可以更高效地探索复杂的分子系统,加速新药和新材料的研发进程。未来,该技术有望实现分子操作的自动化,降低对人工经验的依赖。
📄 摘要(原文)
Molecular dynamics (MD) simulations are a crucial computational tool for researchers to understand and engineer molecular structure and function in areas such as drug discovery, protein engineering, and material design. Despite their utility, MD simulations are expensive, owing to the high dimensionality of molecular systems. Interactive molecular dynamics in virtual reality (iMD-VR) has recently emerged as a "human-in-the-loop" strategy for efficiently navigating hyper-dimensional molecular systems. By providing an immersive 3D environment that enables visualization and manipulation of real-time molecular simulations running on high-performance computing architectures, iMD-VR enables researchers to reach out and guide molecular conformational dynamics, in order to efficiently explore complex, high-dimensional molecular systems. Moreover, iMD-VR simulations generate rich datasets that capture human experts' spatial insight regarding molecular structure and function. This paper explores the use of researcher-generated iMD-VR datasets to train AI agents via imitation learning (IL). IL enables agents to mimic complex behaviours from expert demonstrations, circumventing the need for explicit programming or intricate reward design. In this article, we review IL across robotics and Multi-agents systems domains which are comparable to iMD-VR, and discuss how iMD-VR recordings could be used to train IL models to interact with MD simulations. We then illustrate the applications of these ideas through a proof-of-principle study where iMD-VR data was used to train a CNN network on a simple molecular manipulation task; namely, threading a small molecule through a nanotube pore. Finally, we outline future research directions and potential challenges of using AI agents to augment human expertise in navigating vast molecular conformational spaces.