GP-MoLFormer: A Foundation Model For Molecular Generation
作者: Jerret Ross, Brian Belgodere, Samuel C. Hoffman, Vijil Chenthamarakshan, Jiri Navratil, Youssef Mroueh, Payel Das
分类: q-bio.BM, cs.LG, physics.chem-ph
发布日期: 2024-04-04 (更新: 2025-03-31)
💡 一句话要点
提出GP-MoLFormer以解决分子生成任务的挑战
🎯 匹配领域: 支柱二:RL算法与架构 (RL & Architecture) 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 分子生成 化学语言模型 自回归生成 Transformer 药物发现 材料科学 性质引导优化
📋 核心要点
- 现有的化学语言模型在生成分子时面临训练数据记忆性与新颖性之间的权衡,影响生成效果。
- GP-MoLFormer通过自回归生成和参数高效的微调方法,扩展了化学语言模型在生成任务中的应用。
- 实验结果表明,GP-MoLFormer在去新生成、骨架约束分子装饰和性质引导优化任务上均优于现有基线,展示了其强大的生成能力。
📝 摘要(中文)
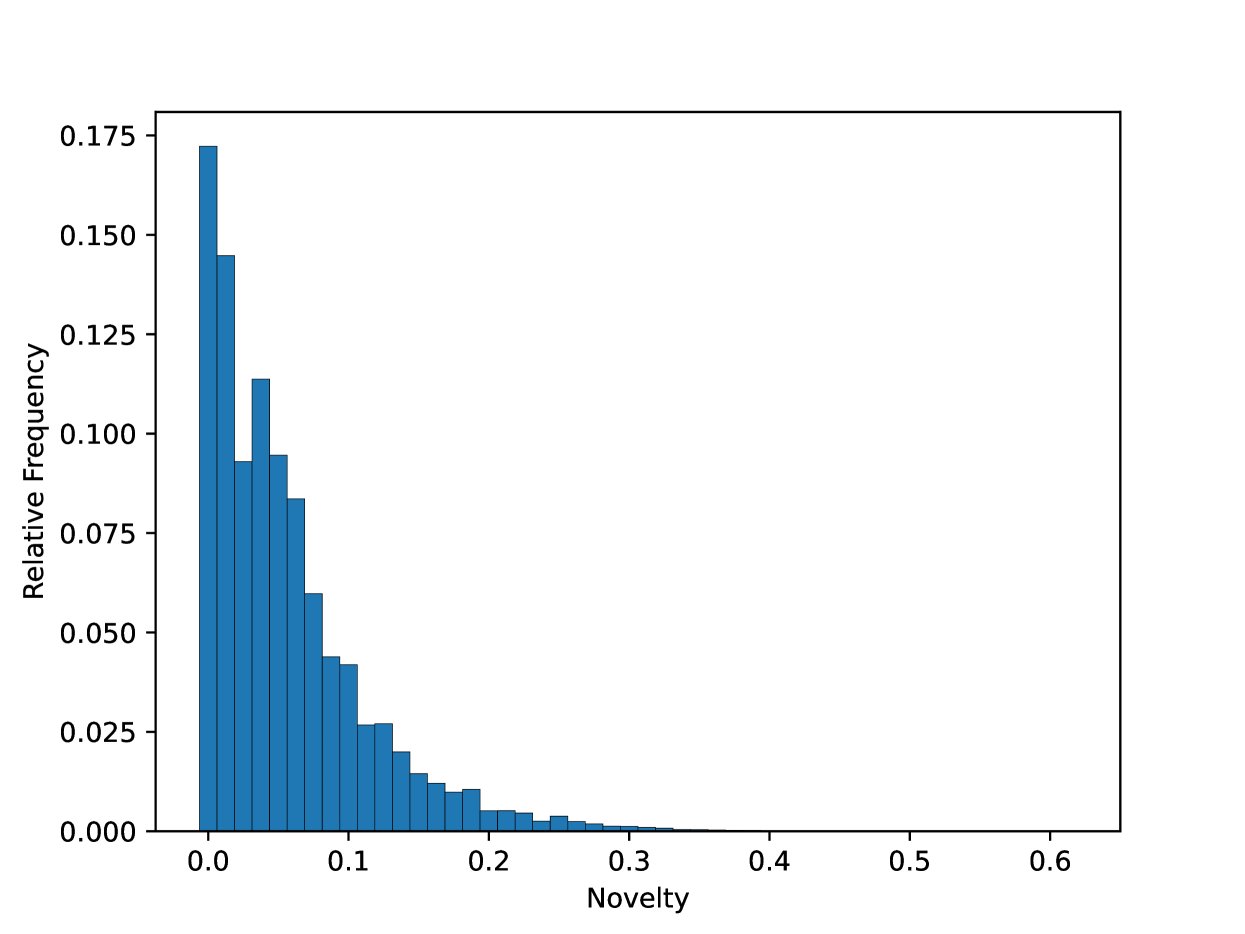
基于Transformer的模型在处理分子字符串的大规模通用数据集上表现出色,成为建模结构-性质关系的强大工具。本文提出GP-MoLFormer,一个自回归的分子字符串生成器,训练于超过11亿个化学SMILES。该模型采用46.8M参数的Transformer解码器,结合线性注意力和旋转位置编码。GP-MoLFormer在去新生成、骨架约束分子装饰和无约束性质引导优化等三项任务上进行了评估,结果显示其在所有任务上均表现优于或与基线相当,展示了其在多种分子生成任务中的广泛适用性。我们还发现GP-MoLFormer在生成中对训练数据的强记忆性,这在化学语言模型中尚未被深入探讨。
🔬 方法详解
问题定义:本文旨在解决分子生成任务中现有方法在训练数据记忆性与生成新颖性之间的矛盾,现有模型在处理大规模化学数据时存在生成效果不理想的问题。
核心思路:GP-MoLFormer通过自回归的方式生成分子字符串,并引入参数高效的微调方法,以提升生成的多样性和质量。该设计旨在充分利用大规模训练数据的潜力,同时避免过度依赖于训练数据。
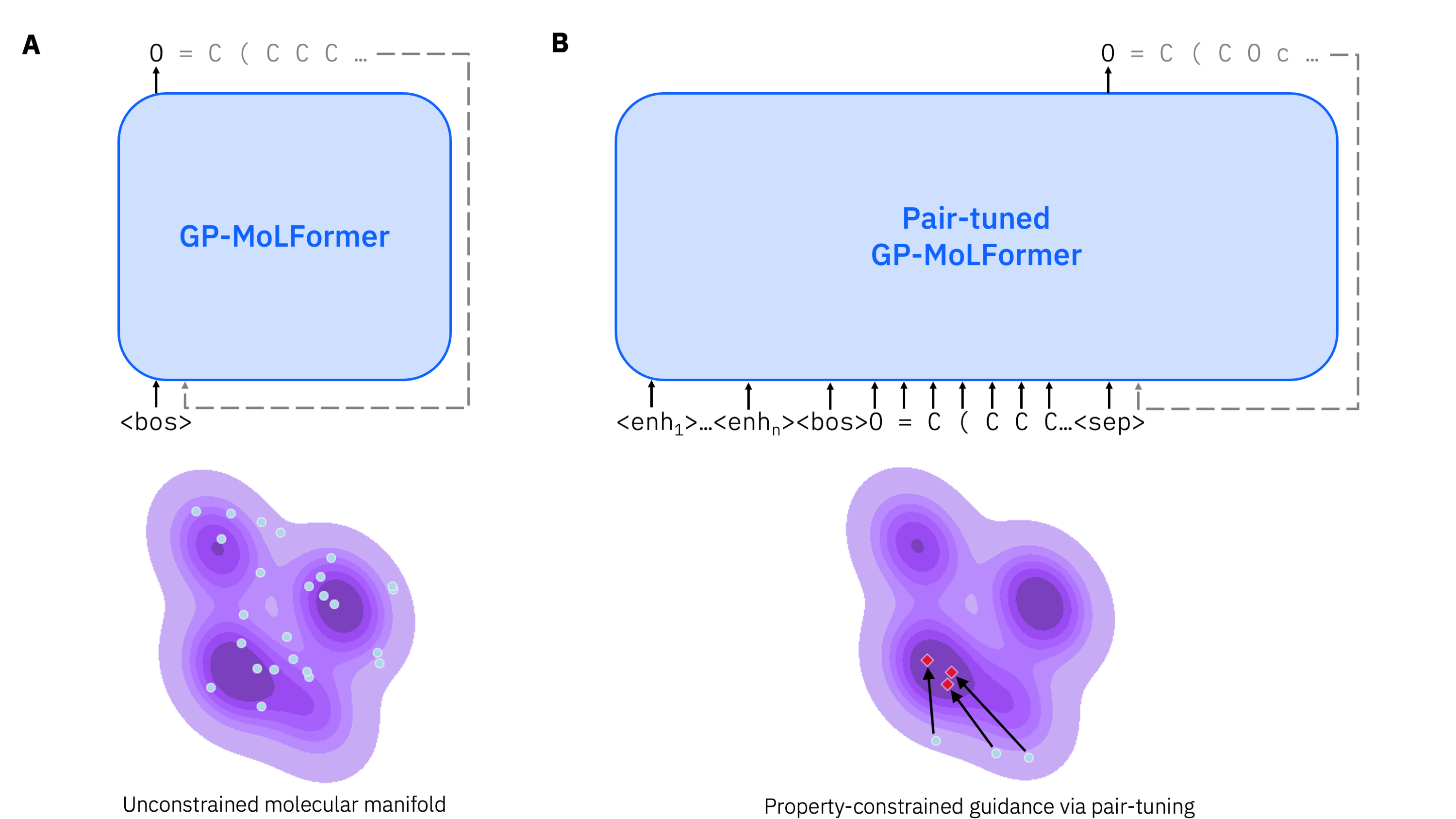
技术框架:GP-MoLFormer的整体架构基于46.8M参数的Transformer解码器,采用线性注意力机制和旋转位置编码。模型在去新生成、骨架约束分子装饰和无约束性质引导优化等任务中进行评估,采用不同的输入策略以适应各类任务需求。
关键创新:最重要的创新在于提出了“对偶微调”方法,通过使用性质有序的分子对作为输入,显著提高了模型在性质引导优化任务中的表现。这一方法在现有文献中尚属首次。
关键设计:模型采用线性注意力机制以降低计算复杂度,旋转位置编码则增强了模型对序列信息的捕捉能力。此外,训练过程中对数据的重复偏差进行了分析,揭示了其对记忆性和新颖性的影响。具体的损失函数和参数设置在实验部分进行了详细说明。
🖼️ 关键图片

📊 实验亮点
实验结果显示,GP-MoLFormer在去新生成和骨架约束分子装饰任务中均优于现有基线,且在性质引导优化任务中通过对偶微调方法显著提升了生成质量。具体而言,模型在所有任务上均表现出色,展示了其在分子生成领域的强大能力。
🎯 应用场景
GP-MoLFormer在药物发现、材料科学等领域具有广泛的应用潜力。其能够高效生成新分子,帮助研究人员快速筛选具有特定性质的化合物,推动新材料和新药物的研发。未来,该模型的进一步优化和扩展可能会在化学领域带来更大的影响。
📄 摘要(原文)
Transformer-based models trained on large and general purpose datasets consisting of molecular strings have recently emerged as a powerful tool for successfully modeling various structure-property relations. Inspired by this success, we extend the paradigm of training chemical language transformers on large-scale chemical datasets to generative tasks in this work. Specifically, we propose GP-MoLFormer, an autoregressive molecular string generator that is trained on more than 1.1B (billion) chemical SMILES. GP-MoLFormer uses a 46.8M parameter transformer decoder model with linear attention and rotary positional encodings as the base architecture. GP-MoLFormer's utility is evaluated and compared with that of existing baselines on three different tasks: de novo generation, scaffold-constrained molecular decoration, and unconstrained property-guided optimization. While the first two are handled with no additional training, we propose a parameter-efficient fine-tuning method for the last task, which uses property-ordered molecular pairs as input. We call this new approach pair-tuning. Our results show GP-MoLFormer performs better or comparable with baselines across all three tasks, demonstrating its general utility for a variety of molecular generation tasks. We further report strong memorization of training data in GP-MoLFormer generations, which has so far remained unexplored for chemical language models. Our analyses reveal that training data memorization and novelty in generations are impacted by the quality and scale of the training data; duplication bias in training data can enhance memorization at the cost of lowering novelty. We further establish a scaling law relating inference compute and novelty in generations.