Integrating Pathology Foundation Models and Spatial Transcriptomics for Cellular Decomposition from Histology Images
作者: Yutong Sun, Sichen Zhu, Peng Qiu
分类: cs.CV
发布日期: 2025-07-09
💡 一句话要点
利用病理学基础模型和空间转录组学,从组织学图像中进行细胞分解
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 病理学基础模型 空间转录组学 细胞分解 组织学图像 多层感知器
📋 核心要点
- 现有方法难以经济高效地从组织学图像中推断细胞组成,限制了对细胞层面病理特征的深入研究。
- 该论文提出利用预训练病理学基础模型提取特征,并用轻量级MLP回归器预测细胞类型丰度,实现高效的细胞分解。
- 实验表明,该方法在预测细胞类型组成方面具有竞争力,同时显著降低了计算复杂度,优于现有方法。
📝 摘要(中文)
数字病理学和深度学习的快速发展促进了病理学基础模型的出现,这些模型有望在各种疾病条件下,通过统一的模型解决一般的病理学问题,无论是否进行微调。与此同时,空间转录组学作为一种变革性技术,能够对苏木精-伊红(H&E)染色的组织学图像进行基因表达谱分析。空间转录组学为在更精细的细胞水平上深入研究现有的组织学图像提供了前所未有的机会。本文提出了一种轻量级且训练高效的方法,通过利用从预训练的病理学基础模型中提取的信息丰富的特征嵌入,直接从H&E染色的组织学图像中预测细胞组成。通过在cell2location衍生的细胞类型丰度上训练一个轻量级多层感知器(MLP)回归器,我们的方法有效地从病理学基础模型中提取知识,并展示了从组织学图像中准确预测细胞类型组成的能力,而无需实际进行昂贵的空间转录组学。与Hist2Cell等现有方法相比,我们的方法表现出具有竞争力的性能,同时显著降低了计算复杂度。
🔬 方法详解
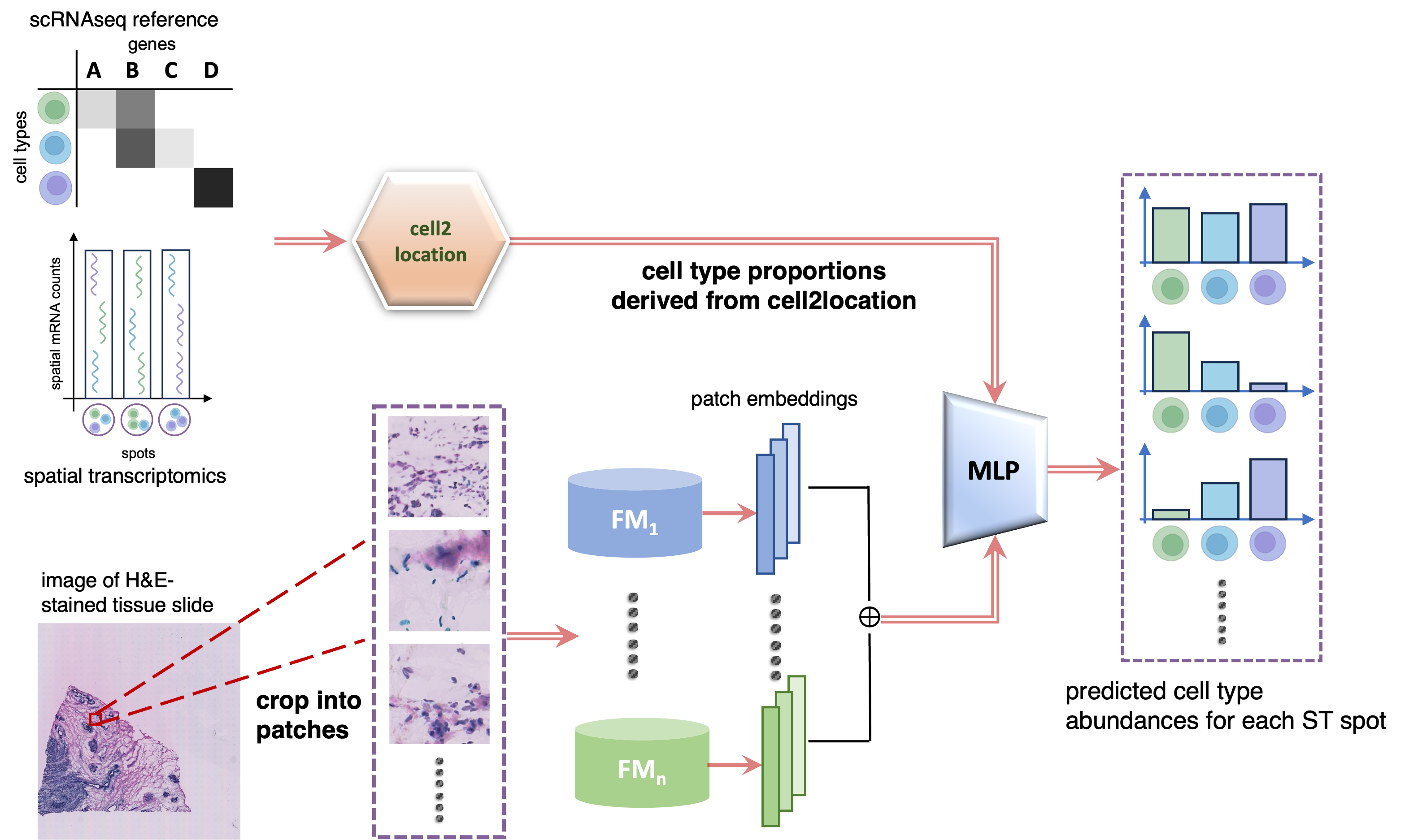
问题定义:论文旨在解决从H&E染色的组织学图像中准确预测细胞类型组成的问题。现有方法,如Hist2Cell,计算复杂度高,需要大量的计算资源和时间,限制了其在实际应用中的可扩展性。此外,直接从图像像素预测细胞组成往往需要大量的标注数据进行训练,成本较高。
核心思路:论文的核心思路是利用预训练的病理学基础模型提取信息丰富的特征嵌入,然后训练一个轻量级的多层感知器(MLP)回归器,将这些特征嵌入映射到细胞类型丰度。这种方法利用了预训练模型学习到的通用病理学知识,从而减少了对大量标注数据的需求,并降低了计算复杂度。
技术框架:整体框架包括以下几个主要阶段:1) 使用预训练的病理学基础模型(如Pathology Foundation Model)提取H&E染色组织学图像的特征嵌入;2) 利用cell2location等方法,从空间转录组学数据中获得细胞类型丰度作为训练标签;3) 使用细胞类型丰度作为监督信号,训练一个轻量级的多层感知器(MLP)回归器,将图像特征嵌入映射到细胞类型丰度;4) 使用训练好的MLP回归器,从新的组织学图像中预测细胞类型组成。
关键创新:该方法最重要的创新点在于利用预训练的病理学基础模型来提取图像特征,从而避免了从头开始训练深度学习模型的需求,显著降低了计算成本和数据需求。此外,使用轻量级的MLP回归器进一步降低了计算复杂度,使其更易于部署和应用。与现有方法相比,该方法在性能上具有竞争力,同时显著降低了计算成本。
关键设计:关键设计包括:1) 选择合适的预训练病理学基础模型,以确保提取的特征能够有效捕捉细胞类型信息;2) 使用cell2location等方法准确估计细胞类型丰度,作为训练标签;3) 设计合适的MLP回归器结构,以平衡模型的表达能力和计算复杂度;4) 使用适当的损失函数(如均方误差)来训练MLP回归器,以最小化预测细胞类型丰度与真实丰度之间的差异。
🖼️ 关键图片


📊 实验亮点
实验结果表明,该方法在预测细胞类型组成方面具有竞争力,与Hist2Cell等现有方法相比,在性能上相当,同时显著降低了计算复杂度。具体而言,该方法能够以更少的计算资源和时间,准确预测组织学图像中各种细胞类型的丰度,为病理学研究和临床应用提供了更高效的工具。
🎯 应用场景
该研究成果可广泛应用于病理学诊断、药物研发和生物学研究等领域。通过从组织学图像中准确预测细胞类型组成,可以辅助病理学家进行疾病诊断和分级,加速药物靶点的发现和验证,并深入理解肿瘤微环境的复杂性。该方法还可以应用于大规模组织学图像分析,为个性化医疗提供更精确的依据。
📄 摘要(原文)
The rapid development of digital pathology and modern deep learning has facilitated the emergence of pathology foundation models that are expected to solve general pathology problems under various disease conditions in one unified model, with or without fine-tuning. In parallel, spatial transcriptomics has emerged as a transformative technology that enables the profiling of gene expression on hematoxylin and eosin (H&E) stained histology images. Spatial transcriptomics unlocks the unprecedented opportunity to dive into existing histology images at a more granular, cellular level. In this work, we propose a lightweight and training-efficient approach to predict cellular composition directly from H&E-stained histology images by leveraging information-enriched feature embeddings extracted from pre-trained pathology foundation models. By training a lightweight multi-layer perceptron (MLP) regressor on cell-type abundances derived via cell2location, our method efficiently distills knowledge from pathology foundation models and demonstrates the ability to accurately predict cell-type compositions from histology images, without physically performing the costly spatial transcriptomics. Our method demonstrates competitive performance compared to existing methods such as Hist2Cell, while significantly reducing computational complexity.