MD-LLM-1: A Large Language Model for Molecular Dynamics
作者: Mhd Hussein Murtada, Z. Faidon Brotzakis, Michele Vendruscolo
分类: q-bio.BM, cs.CL, cs.LG, physics.comp-ph
发布日期: 2025-07-21
💡 一句话要点
提出MD-LLM-1,利用大语言模型学习蛋白质动力学并预测构象状态
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 分子动力学 大语言模型 蛋白质动力学 构象预测 深度学习
📋 核心要点
- 分子动力学模拟计算量大,限制了其在生物大分子系统中的应用。
- 提出MD-LLM框架,利用大语言模型学习蛋白质动力学,预测未见构象。
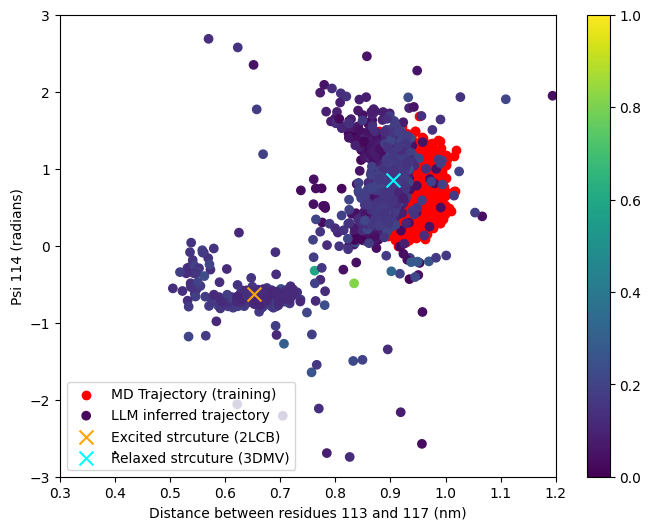
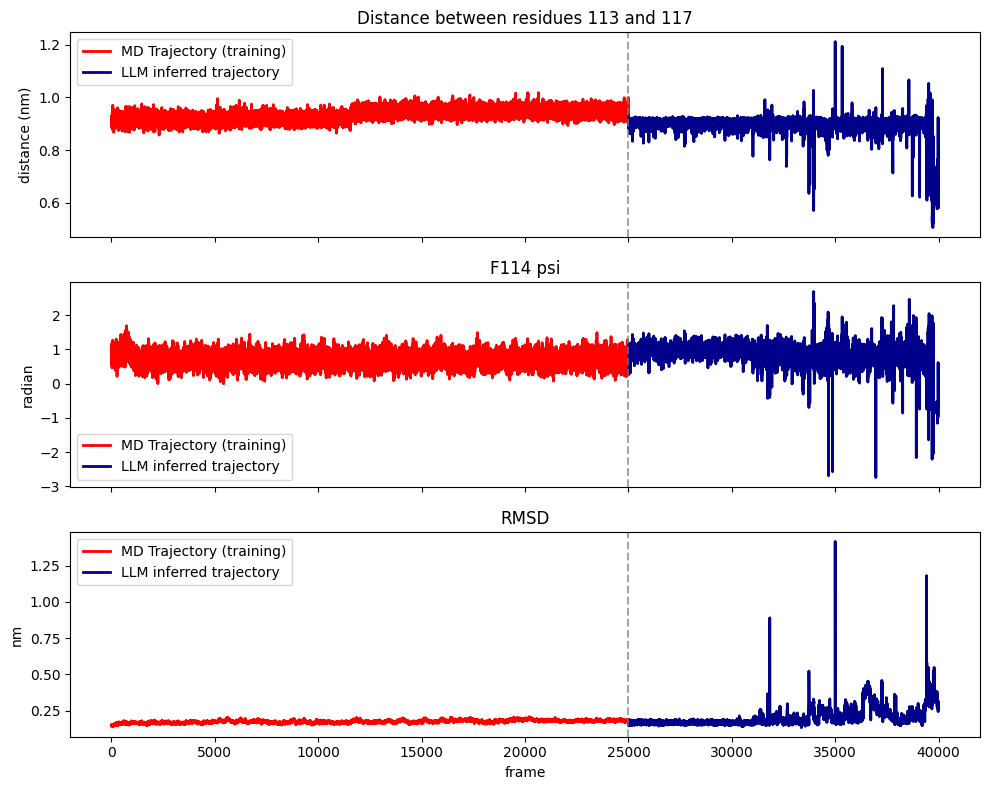
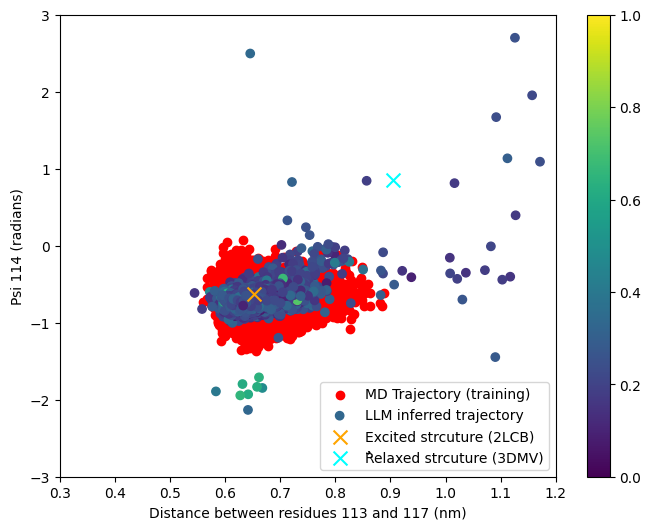
- MD-LLM-1在T4溶菌酶和Mad2蛋白上验证,可基于单一构象预测其他构象。
📝 摘要(中文)
分子动力学(MD)是模拟分子系统的强大方法,但对于许多具有生物学意义的大分子系统,其在空间和时间尺度上的计算量仍然很大。为了探索深度学习解决此问题的机会,我们引入了一个分子动力学大语言模型(MD-LLM)框架,以说明如何利用LLM来学习蛋白质动力学并发现训练中未见的状态。通过将MD-LLM-1(该方法的第一个实现,通过微调Mistral 7B获得)应用于T4溶菌酶和Mad2蛋白系统,我们表明,在一个构象状态上进行训练能够预测其他构象状态。这些结果表明,MD-LLM-1可以学习探索蛋白质构象景观的原理,尽管它尚未明确地模拟其热力学和动力学。
🔬 方法详解
问题定义:分子动力学模拟在研究生物大分子系统时,由于其巨大的计算量,难以在合理的时间内模拟足够长的轨迹,从而限制了对蛋白质构象空间的探索。现有方法难以有效学习蛋白质的复杂动力学行为,尤其是在探索未知的构象状态方面存在挑战。
核心思路:该论文的核心思路是利用大语言模型(LLM)强大的序列建模能力,将蛋白质的动力学轨迹视为一种“语言”,通过训练LLM来学习蛋白质构象之间的关系和转换规则。通过这种方式,LLM可以预测蛋白质在不同时间步长的构象,从而加速分子动力学模拟,并发现新的构象状态。
技术框架:MD-LLM框架包含以下主要步骤:1) 数据准备:将分子动力学模拟产生的蛋白质轨迹数据进行预处理,例如将原子坐标转换为适合LLM处理的序列数据。2) 模型选择与微调:选择一个预训练的LLM(例如Mistral 7B),并使用准备好的蛋白质轨迹数据进行微调,得到MD-LLM-1。3) 构象预测:使用训练好的MD-LLM-1,输入初始构象,预测后续时间步长的构象序列。4) 结果分析:分析预测的构象序列,评估模型的性能,并探索新的构象状态。
关键创新:该论文的关键创新在于将大语言模型应用于分子动力学模拟领域,提出了一种新的蛋白质动力学建模方法。与传统的分子动力学方法相比,MD-LLM-1能够学习蛋白质构象之间的复杂关系,并预测未知的构象状态,从而加速模拟过程,并扩展了对蛋白质构象空间的探索能力。
关键设计:MD-LLM-1基于Mistral 7B进行微调,具体的技术细节包括:选择合适的训练数据集,设计合适的损失函数(例如,均方误差损失函数),调整模型的超参数(例如,学习率、batch size),以及采用适当的训练策略(例如,early stopping)。论文中还提到,该模型尚未明确地模拟蛋白质的热力学和动力学性质,这可能是未来研究的一个方向。
🖼️ 关键图片
📊 实验亮点
MD-LLM-1在T4溶菌酶和Mad2蛋白系统上的实验结果表明,该模型能够学习蛋白质的构象景观,并基于单一构象预测其他构象。虽然论文中没有给出具体的性能数据,但实验结果验证了MD-LLM框架的有效性,并为未来的研究奠定了基础。该模型能够探索蛋白质的构象空间,即使没有明确模拟热力学和动力学,也展示了其潜力。
🎯 应用场景
MD-LLM框架可应用于蛋白质结构预测、药物发现、蛋白质工程等领域。通过加速分子动力学模拟,可以更高效地研究蛋白质的构象变化、相互作用以及与配体的结合,从而为药物设计提供更准确的依据。此外,该方法还可以用于预测蛋白质突变体的结构和功能,加速蛋白质工程的进程。
📄 摘要(原文)
Molecular dynamics (MD) is a powerful approach for modelling molecular systems, but it remains computationally intensive on spatial and time scales of many macromolecular systems of biological interest. To explore the opportunities offered by deep learning to address this problem, we introduce a Molecular Dynamics Large Language Model (MD-LLM) framework to illustrate how LLMs can be leveraged to learn protein dynamics and discover states not seen in training. By applying MD-LLM-1, the first implementation of this approach, obtained by fine-tuning Mistral 7B, to the T4 lysozyme and Mad2 protein systems, we show that training on one conformational state enables the prediction of other conformational states. These results indicate that MD-LLM-1 can learn the principles for the exploration of the conformational landscapes of proteins, although it is not yet modeling explicitly their thermodynamics and kinetics.