Bidirectional Mamba for Single-Cell Data: Efficient Context Learning with Biological Fidelity
作者: Cong Qi, Hanzhang Fang, Tianxing Hu, Siqi Jiang, Wei Zhi
分类: cs.CL, cs.LG, q-bio.GN
发布日期: 2025-04-22 (更新: 2025-12-26)
💡 一句话要点
GeneMamba:基于双向Mamba的单细胞数据高效上下文学习模型
🎯 匹配领域: 支柱二:RL算法与架构 (RL & Architecture) 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 单细胞RNA测序 状态空间模型 Mamba Bi-Mamba 基因上下文学习 生物信息学 深度学习
📋 核心要点
- 单细胞RNA测序数据具有高维度、稀疏性和批次效应等复杂性,对计算方法提出了挑战,Transformer模型存在二次复杂度问题。
- GeneMamba利用Bi-Mamba架构,以线性时间复杂度捕获双向基因上下文,显著提升计算效率,并结合生物信息进行预训练。
- 实验结果表明,GeneMamba在多批次整合、细胞类型注释和基因-基因相关性等任务上表现出强大的性能、可解释性和鲁棒性。
📝 摘要(中文)
本文提出GeneMamba,一个基于状态空间模型的、可扩展且高效的单细胞转录组学基础模型。GeneMamba利用Bi-Mamba架构,以线性时间复杂度捕获双向基因上下文信息,相比于Transformer模型,在计算效率上有显著提升。该模型在近3000万个细胞上进行预训练,并结合了生物信息驱动的目标,包括通路感知的对比损失和基于排序的基因编码。在多批次整合、细胞类型注释和基因-基因相关性等多种任务上的评估表明,GeneMamba具有强大的性能、可解释性和鲁棒性。这些结果表明,GeneMamba是Transformer模型的实用且强大的替代方案,推动了生物学基础的、可扩展的单细胞数据分析工具的发展。
🔬 方法详解
问题定义:单细胞RNA测序数据的分析面临高维度、稀疏性和批次效应等挑战。现有的Transformer模型虽然在处理序列数据方面表现出色,但其计算复杂度是二次的,难以扩展到大规模单细胞数据集,并且在捕捉长程依赖关系方面存在局限性。
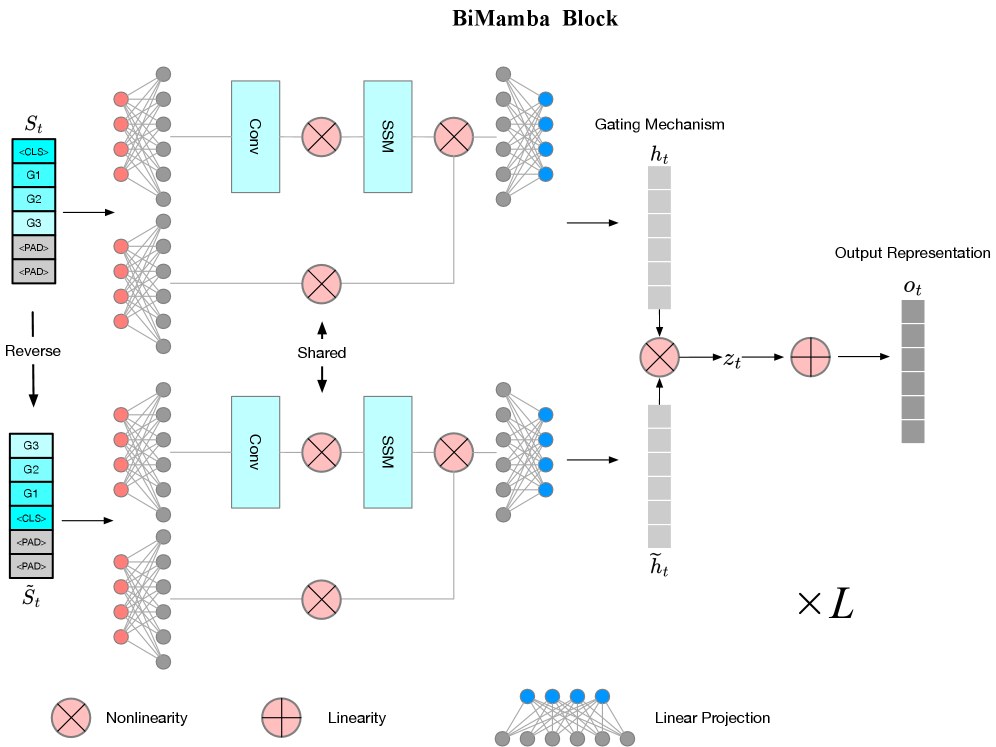
核心思路:GeneMamba的核心思路是利用状态空间模型(SSM)中的Mamba架构,特别是Bi-Mamba,来高效地建模基因之间的依赖关系。Mamba具有线性时间复杂度,能够处理长序列数据,并且通过选择性扫描机制,能够更好地捕捉上下文信息。通过双向建模,GeneMamba可以同时考虑基因的上游和下游信息,从而更全面地理解基因之间的相互作用。
技术框架:GeneMamba的整体框架包括数据预处理、模型构建、预训练和下游任务微调等阶段。首先,对单细胞RNA测序数据进行标准化和批次校正。然后,构建基于Bi-Mamba的神经网络模型,该模型由多个Mamba块组成。在预训练阶段,使用大规模单细胞数据集,并结合生物信息驱动的目标函数进行训练。最后,在下游任务中,对预训练模型进行微调,以适应特定的任务需求。
关键创新:GeneMamba的关键创新在于以下几个方面:1) 采用Bi-Mamba架构,实现了线性时间复杂度的基因上下文建模;2) 结合生物信息,设计了通路感知的对比损失和基于排序的基因编码,从而提高了模型的生物学合理性;3) 在大规模单细胞数据集上进行预训练,使得模型具有更好的泛化能力。与Transformer模型相比,GeneMamba在计算效率和长程依赖关系建模方面具有显著优势。
关键设计:在模型设计方面,GeneMamba采用了多层Bi-Mamba块堆叠的结构,每个Mamba块包含选择性扫描机制和状态空间模型。通路感知的对比损失旨在拉近同一通路的基因表达谱,而推远不同通路的基因表达谱。基于排序的基因编码将基因表达值转换为排序值,从而减少了批次效应的影响。预训练阶段使用了AdamW优化器,并设置了合适的学习率和权重衰减系数。
🖼️ 关键图片


📊 实验亮点
GeneMamba在多批次整合、细胞类型注释和基因-基因相关性等任务上取得了显著的性能提升。例如,在细胞类型注释任务中,GeneMamba的准确率超过了现有的Transformer模型和其他单细胞分析方法。此外,GeneMamba还展现出良好的可解释性,能够识别与特定细胞类型或疾病相关的关键基因和通路。
🎯 应用场景
GeneMamba可应用于多种单细胞数据分析任务,包括细胞类型注释、基因调控网络推断、疾病机制研究和药物靶点发现等。通过高效地建模基因之间的复杂关系,GeneMamba能够帮助研究人员更深入地理解细胞异质性和疾病发生发展的分子机制,从而为精准医疗提供新的思路和方法。
📄 摘要(原文)
Single-cell RNA sequencing (scRNA-seq) enables high-resolution analysis of cellular heterogeneity, but its complexity, which is marked by high dimensionality, sparsity, and batch effects, which poses major computational challenges. Transformer-based models have made significant advances in this domain but are often limited by their quadratic complexity and suboptimal handling of long-range dependencies. In this work, we introduce GeneMamba, a scalable and efficient foundation model for single-cell transcriptomics built on state space modeling. Leveraging the Bi-Mamba architecture, GeneMamba captures bidirectional gene context with linear-time complexity, offering substantial computational gains over transformer baselines. The model is pretrained on nearly 30 million cells and incorporates biologically informed objectives, including pathway-aware contrastive loss and rank-based gene encoding. We evaluate GeneMamba across diverse tasks, including multi-batch integration, cell type annotation, and gene-gene correlation, demonstrating strong performance, interpretability, and robustness. These results position GeneMamba as a practical and powerful alternative to transformer-based methods, advancing the development of biologically grounded, scalable tools for large-scale single-cell data analysis.