PathoLM: Identifying pathogenicity from the DNA sequence through the Genome Foundation Model
作者: Sajib Acharjee Dip, Uddip Acharjee Shuvo, Tran Chau, Haoqiu Song, Petra Choi, Xuan Wang, Liqing Zhang
分类: cs.CL, cs.LG, q-bio.GN
发布日期: 2024-06-19
备注: 9 pages, 3 figures
💡 一句话要点
PathoLM:利用基因组基础模型从DNA序列中识别病原体
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 病原体识别 基因组基础模型 DNA序列 深度学习 预训练模型 零样本学习 小样本学习 ESKAPEE病原体
📋 核心要点
- 传统病原体识别方法计算量大,依赖参考数据库,对新型病原体检测效果不佳,传统机器学习方法易过拟合。
- PathoLM利用预训练DNA模型,通过少量数据微调,捕获更广泛的基因组上下文,提升新型病原体识别能力。
- 实验结果表明,PathoLM在零样本和小样本学习中优于现有模型,并在ESKAPEE物种分类中表现出色。
📝 摘要(中文)
病原体识别对于疾病的诊断、治疗和预防至关重要,对控制感染和保障公共健康起着关键作用。传统的基于比对的方法虽然应用广泛,但计算密集,依赖于广泛的参考数据库,并且由于其低灵敏度和特异性,常常无法检测到新的病原体。 传统的机器学习技术虽然有前景,但需要大量的带注释数据集和广泛的特征工程,并且容易过拟合。 为了解决这些挑战,我们引入了PathoLM,这是一个先进的病原体语言模型,针对细菌和病毒序列中的致病性识别进行了优化。 PathoLM利用了诸如Nucleotide Transformer之类的预训练DNA模型的优势,只需要最少量的数据进行微调,从而增强了病原体检测能力。 它有效地捕获了更广泛的基因组上下文,从而显着改善了新型和不同病原体的识别。 我们开发了一个包含大约30种病毒和细菌(包括ESKAPEE病原体)的综合数据集,其中包括七种对抗生素具有抗药性的高毒力细菌菌株。 此外,我们还整理了一个专门针对ESKAPEE组的物种分类数据集。 在比较评估中,PathoLM显着优于诸如DciPatho之类的现有模型,从而证明了强大的零样本和小样本能力。 此外,我们扩展了PathoLM-Sp用于ESKAPEE物种分类,尽管任务复杂,但与其他先进的深度学习方法相比,它表现出卓越的性能。
🔬 方法详解
问题定义:论文旨在解决病原体识别问题,特别是针对新型和变异病原体,传统方法如基于比对的方法计算成本高昂,依赖大量参考数据库,且对新型病原体识别能力有限。传统的机器学习方法需要大量标注数据,容易过拟合,泛化能力不足。
核心思路:论文的核心思路是利用预训练的基因组基础模型(Genome Foundation Model),特别是Nucleotide Transformer,通过少量数据进行微调,从而实现高效且准确的病原体识别。这种方法能够捕获更广泛的基因组上下文信息,提高对新型和变异病原体的识别能力。
技术框架:PathoLM的技术框架主要包括以下几个阶段:1) 使用大规模基因组数据预训练基础模型(如Nucleotide Transformer);2) 构建包含多种病毒和细菌(包括ESKAPEE病原体)的综合数据集;3) 使用少量标注数据对预训练模型进行微调,得到PathoLM模型;4) 在病原体识别和物种分类任务上评估PathoLM的性能。
关键创新:PathoLM的关键创新在于利用基因组基础模型进行病原体识别,这与传统的基于比对或特征工程的方法有本质区别。通过预训练,模型能够学习到基因组的通用表示,从而在少量数据下也能实现良好的泛化能力。此外,PathoLM能够捕获更广泛的基因组上下文信息,有助于识别新型和变异病原体。
关键设计:论文中关键的设计包括:1) 选择Nucleotide Transformer作为基础模型,因为它在处理DNA序列方面表现出色;2) 构建包含多种病原体的数据集,以提高模型的泛化能力;3) 使用少量数据进行微调,以降低标注成本;4) 针对ESKAPEE物种分类任务,扩展了PathoLM-Sp模型,并进行了专门的优化。
🖼️ 关键图片


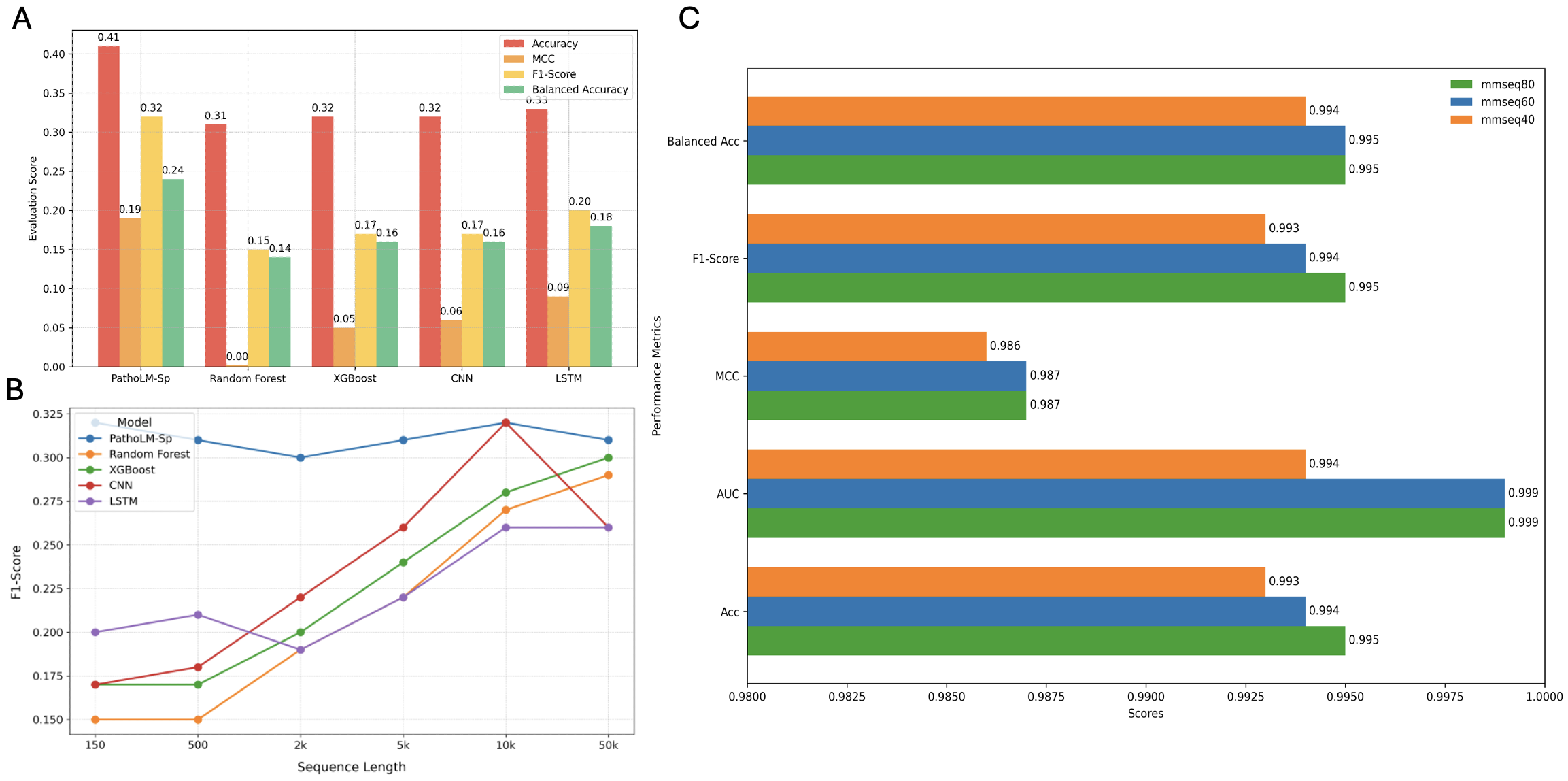
📊 实验亮点
PathoLM在病原体识别任务中表现出色,显著优于现有模型DciPatho,展现出强大的零样本和小样本学习能力。在ESKAPEE物种分类任务中,PathoLM-Sp模型也超越了其他先进的深度学习方法。这些结果表明,PathoLM能够有效利用基因组基础模型进行病原体识别,具有很高的实用价值。
🎯 应用场景
PathoLM具有广泛的应用前景,可用于疾病诊断、疫情监控、新药研发等领域。通过快速准确地识别病原体,可以帮助医生制定更有效的治疗方案,控制疫情蔓延。此外,PathoLM还可以用于发现新的病原体,为新药研发提供线索。该研究有望推动病原体识别技术的发展,为公共卫生安全做出贡献。
📄 摘要(原文)
Pathogen identification is pivotal in diagnosing, treating, and preventing diseases, crucial for controlling infections and safeguarding public health. Traditional alignment-based methods, though widely used, are computationally intense and reliant on extensive reference databases, often failing to detect novel pathogens due to their low sensitivity and specificity. Similarly, conventional machine learning techniques, while promising, require large annotated datasets and extensive feature engineering and are prone to overfitting. Addressing these challenges, we introduce PathoLM, a cutting-edge pathogen language model optimized for the identification of pathogenicity in bacterial and viral sequences. Leveraging the strengths of pre-trained DNA models such as the Nucleotide Transformer, PathoLM requires minimal data for fine-tuning, thereby enhancing pathogen detection capabilities. It effectively captures a broader genomic context, significantly improving the identification of novel and divergent pathogens. We developed a comprehensive data set comprising approximately 30 species of viruses and bacteria, including ESKAPEE pathogens, seven notably virulent bacterial strains resistant to antibiotics. Additionally, we curated a species classification dataset centered specifically on the ESKAPEE group. In comparative assessments, PathoLM dramatically outperforms existing models like DciPatho, demonstrating robust zero-shot and few-shot capabilities. Furthermore, we expanded PathoLM-Sp for ESKAPEE species classification, where it showed superior performance compared to other advanced deep learning methods, despite the complexities of the task.