DrugLLM: Open Large Language Model for Few-shot Molecule Generation
作者: Xianggen Liu, Yan Guo, Haoran Li, Jin Liu, Shudong Huang, Bowen Ke, Jiancheng Lv
分类: q-bio.BM, cs.CL, cs.LG
发布日期: 2024-05-07
备注: 17 pages, 3 figures
💡 一句话要点
DrugLLM:用于少样本分子生成的开放大型语言模型
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 药物设计 大型语言模型 少样本学习 分子生成 分子表示
📋 核心要点
- 现有大型语言模型在生物和化学语言处理方面存在不足,难以捕捉分子结构与药理性质间的关系。
- DrugLLM采用基于组的分子表示(GMR)将分子排列成序列,学习分子修饰以增强特定性质。
- 实验表明,DrugLLM能够基于少量示例生成具有预期性质的新分子,展示了强大的少样本分子生成能力。
📝 摘要(中文)
大型语言模型(LLM)在语言处理和计算机视觉等领域取得了显著进展。尽管涌现出各种提高少样本学习能力的技术,但当前的LLM在处理生物和化学语言方面仍然不足。例如,它们难以捕捉分子结构和药理化学性质之间的关系。因此,小分子药物修饰的少样本学习能力仍然受到阻碍。在这项工作中,我们引入了DrugLLM,这是一个为药物设计量身定制的LLM。在训练过程中,我们采用基于组的分子表示(GMR)来表示分子,并将它们排列成序列,以反映旨在增强特定分子性质的修饰。DrugLLM通过预测基于过去修饰的下一个分子来学习如何在药物发现中修饰分子。大量的计算实验表明,DrugLLM可以基于有限的例子生成具有预期性质的新分子,从而展现出强大的少样本分子生成能力。
🔬 方法详解
问题定义:论文旨在解决现有大型语言模型在小分子药物设计中,少样本学习能力不足的问题。现有方法难以有效捕捉分子结构与药理性质之间的复杂关系,导致药物修饰效率低下。
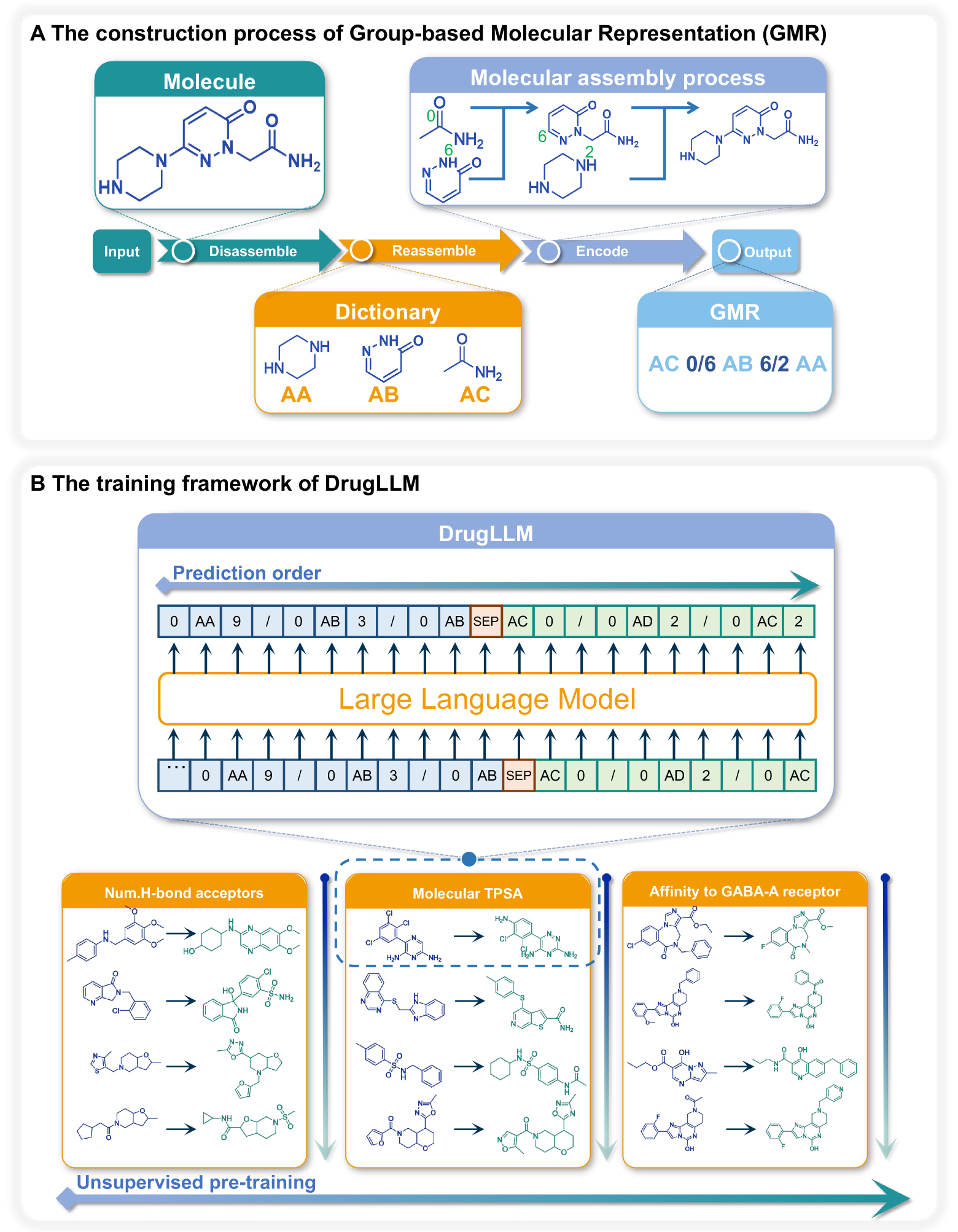
核心思路:论文的核心思路是构建一个专门为药物设计定制的大型语言模型DrugLLM。通过在训练过程中引入基于组的分子表示(GMR),使模型能够学习分子修饰与性质增强之间的关联,从而提升少样本分子生成能力。
技术框架:DrugLLM的整体框架包括数据预处理、模型训练和分子生成三个主要阶段。首先,利用GMR对分子进行序列化表示。然后,使用大量药物分子数据对LLM进行训练,使其学习分子修饰的模式。最后,在少样本条件下,DrugLLM能够根据给定的分子和期望性质,生成新的分子。
关键创新:该论文的关键创新在于提出了基于组的分子表示(GMR)方法,将分子结构信息编码成序列,使得LLM能够更好地理解和学习分子之间的关系。此外,DrugLLM是首个专门为药物设计定制的LLM,针对性地解决了药物发现领域的特定问题。
关键设计:GMR的具体实现方式未知,但其核心思想是将分子分解为不同的功能组,并按照一定的规则进行排序,形成序列。损失函数的设计目标是使模型能够准确预测下一个分子,从而学习到分子修饰的规律。具体的网络结构细节未知,但可以推测使用了Transformer等常用的LLM架构。
🖼️ 关键图片


📊 实验亮点
论文通过大量计算实验验证了DrugLLM的有效性。实验结果表明,DrugLLM能够基于少量示例生成具有预期性质的新分子,展现出强大的少样本分子生成能力。具体的性能数据和对比基线未知,但论文强调了DrugLLM在少样本学习方面的优势。
🎯 应用场景
DrugLLM在药物发现领域具有广泛的应用前景,可以用于先导化合物的优化、新药设计和药物重定位等任务。通过少样本学习,DrugLLM能够快速生成具有特定性质的候选药物分子,加速药物研发进程,降低研发成本。未来,DrugLLM有望成为药物研发人员的重要工具,推动药物创新。
📄 摘要(原文)
Large Language Models (LLMs) have made great strides in areas such as language processing and computer vision. Despite the emergence of diverse techniques to improve few-shot learning capacity, current LLMs fall short in handling the languages in biology and chemistry. For example, they are struggling to capture the relationship between molecule structure and pharmacochemical properties. Consequently, the few-shot learning capacity of small-molecule drug modification remains impeded. In this work, we introduced DrugLLM, a LLM tailored for drug design. During the training process, we employed Group-based Molecular Representation (GMR) to represent molecules, arranging them in sequences that reflect modifications aimed at enhancing specific molecular properties. DrugLLM learns how to modify molecules in drug discovery by predicting the next molecule based on past modifications. Extensive computational experiments demonstrate that DrugLLM can generate new molecules with expected properties based on limited examples, presenting a powerful few-shot molecule generation capacity.