Integrating Large Language Models for Genetic Variant Classification
作者: Youssef Boulaimen, Gabriele Fossi, Leila Outemzabet, Nathalie Jeanray, Oleksandr Levenets, Stephane Gerart, Sebastien Vachenc, Salvatore Raieli, Joanna Giemza
分类: q-bio.GN, cs.AI, cs.LG
发布日期: 2024-11-07
备注: 21 pages, 7 figures
💡 一句话要点
整合大型语言模型以提升遗传变异分类的准确性和可靠性
🎯 匹配领域: 支柱九:具身大模型 (Embodied Foundation Models)
关键词: 遗传变异分类 大型语言模型 精准医学 蛋白质序列 DNA序列
📋 核心要点
- 现有遗传变异分类方法在处理意义未明变异(VUS)时面临挑战,准确性有待提高。
- 本研究整合GPN-MSA、ESM1b和AlphaMissense等LLM,利用DNA、蛋白质序列和结构信息进行综合分析。
- 实验结果表明,该方法在ProteinGym和ClinVar数据集上显著提升了变异分类的准确性和可靠性。
📝 摘要(中文)
遗传变异的分类,特别是意义未明变异(VUS)的分类,是临床遗传学和精准医学中的一项重大挑战。大型语言模型(LLM)已成为该领域变革性的工具。这些模型可以揭示传统方法可能遗漏的复杂模式和预测性见解,从而提高遗传变异致病性的预测准确性。本研究探讨了最先进的LLM的整合,包括GPN-MSA、ESM1b和AlphaMissense,它们利用DNA和蛋白质序列数据以及结构见解,形成一个全面的变异分类分析框架。我们的方法使用注释良好的ProteinGym和ClinVar数据集评估这些集成模型,在分类性能方面设定了新的基准。这些模型在一组具有挑战性的变异上进行了严格的测试,证明了相对于现有最先进工具的显著改进,尤其是在处理模糊和临床上不确定的变异方面。这项研究的结果强调了结合多种建模方法以显著提高遗传变异分类系统的准确性和可靠性的有效性。这些发现支持在临床环境中部署这些先进的计算模型,它们可以显著增强遗传疾病的诊断过程,最终通过提供更详细和可操作的遗传见解来推动个性化医疗的边界。
🔬 方法详解
问题定义:论文旨在解决遗传变异分类,特别是意义未明变异(VUS)分类的准确性问题。现有方法在处理复杂和模糊的变异时表现不足,导致临床诊断和个性化医疗的挑战。传统方法可能无法充分利用DNA和蛋白质序列数据中的复杂模式和结构信息。
核心思路:论文的核心思路是整合多个大型语言模型(LLM),利用它们各自的优势,形成一个综合的分析框架。通过结合GPN-MSA、ESM1b和AlphaMissense等模型的预测结果,可以更全面地评估变异的致病性。这种集成方法旨在克服单一模型的局限性,提高分类的准确性和鲁棒性。
技术框架:该研究的技术框架主要包括以下几个阶段:1) 数据准备:收集和预处理遗传变异数据,包括DNA和蛋白质序列信息,以及结构数据。2) 模型集成:选择并集成多个LLM,包括GPN-MSA、ESM1b和AlphaMissense。3) 预测:使用集成模型对遗传变异的致病性进行预测。4) 评估:使用ProteinGym和ClinVar等数据集评估模型的性能,并与现有方法进行比较。
关键创新:该研究的关键创新在于整合了多个LLM,形成一个综合的变异分类框架。与传统的单一模型方法相比,这种集成方法能够更全面地利用各种信息源,从而提高分类的准确性和可靠性。此外,该研究还探索了如何有效地结合不同模型的预测结果,以获得最佳的性能。
关键设计:具体的技术细节包括:1) LLM的选择:选择GPN-MSA、ESM1b和AlphaMissense等模型,是因为它们在处理DNA和蛋白质序列数据方面具有优势。2) 数据集:使用ProteinGym和ClinVar等数据集进行评估,这些数据集包含大量的遗传变异信息,并经过了人工标注。3) 评估指标:使用准确率、召回率、F1值等指标评估模型的性能。4) 模型集成方法:研究了不同的模型集成方法,例如加权平均、投票等,以获得最佳的性能。
🖼️ 关键图片

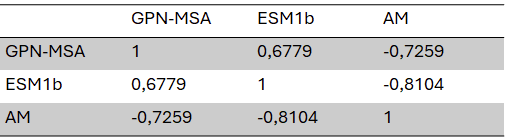

📊 实验亮点
该研究在ProteinGym和ClinVar数据集上进行了实验,结果表明,集成模型在变异分类的准确性和可靠性方面显著优于现有方法。特别是在处理模糊和临床上不确定的变异方面,该方法表现出明显的优势。具体性能数据未知,但摘要强调了“substantial improvements over existing state-of-the-art tools”。
🎯 应用场景
该研究成果可应用于临床遗传学、精准医学和药物研发等领域。通过提高遗传变异分类的准确性,可以帮助医生更准确地诊断遗传疾病,为患者提供更个性化的治疗方案。此外,该研究还可以加速药物研发过程,帮助科学家发现新的药物靶点。
📄 摘要(原文)
The classification of genetic variants, particularly Variants of Uncertain Significance (VUS), poses a significant challenge in clinical genetics and precision medicine. Large Language Models (LLMs) have emerged as transformative tools in this realm. These models can uncover intricate patterns and predictive insights that traditional methods might miss, thus enhancing the predictive accuracy of genetic variant pathogenicity. This study investigates the integration of state-of-the-art LLMs, including GPN-MSA, ESM1b, and AlphaMissense, which leverage DNA and protein sequence data alongside structural insights to form a comprehensive analytical framework for variant classification. Our approach evaluates these integrated models using the well-annotated ProteinGym and ClinVar datasets, setting new benchmarks in classification performance. The models were rigorously tested on a set of challenging variants, demonstrating substantial improvements over existing state-of-the-art tools, especially in handling ambiguous and clinically uncertain variants. The results of this research underline the efficacy of combining multiple modeling approaches to significantly refine the accuracy and reliability of genetic variant classification systems. These findings support the deployment of these advanced computational models in clinical environments, where they can significantly enhance the diagnostic processes for genetic disorders, ultimately pushing the boundaries of personalized medicine by offering more detailed and actionable genetic insights.